Journal of Structural Biology ( IF 3.0 ) Pub Date : 2020-09-10 , DOI: 10.1016/j.jsb.2020.107617 Konstantina Karathanou 1 , Michalis Lazaratos 1 , Éva Bertalan 1 , Malte Siemers 1 , Krzysztof Buzar 1 , Gebhard F X Schertler 2 , Coral Del Val 3 , Ana-Nicoleta Bondar 1
|
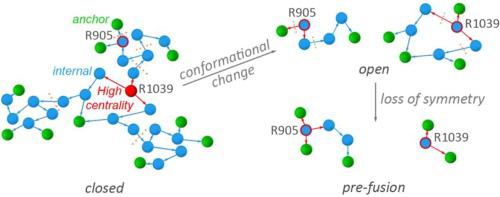
Corona virus spike protein S is a large homo-trimeric protein anchored in the membrane of the virion particle. Protein S binds to angiotensin-converting-enzyme 2, ACE2, of the host cell, followed by proteolysis of the spike protein, drastic protein conformational change with exposure of the fusion peptide of the virus, and entry of the virion into the host cell. The structural elements that govern conformational plasticity of the spike protein are largely unknown. Here, we present a methodology that relies upon graph and centrality analyses, augmented by bioinformatics, to identify and characterize large H-bond clusters in protein structures. We apply this methodology to protein S ectodomain and find that, in the closed conformation, the three protomers of protein S bring the same contribution to an extensive central network of H-bonds, and contribute symmetrically to a relatively large H-bond cluster at the receptor binding domain, and to a cluster near a protease cleavage site. Markedly different H-bonding at these three clusters in open and pre-fusion conformations suggest dynamic H-bond clusters could facilitate structural plasticity and selection of a protein S protomer for binding to the host receptor, and proteolytic cleavage. From analyses of spike protein sequences we identify patches of histidine and carboxylate groups that could be involved in transient proton binding.
中文翻译:
基于图的方法识别了 SARS-CoV-2 刺突蛋白 S 中的动态氢键通信网络。
冠状病毒刺突蛋白 S 是一种锚定在病毒颗粒膜上的大型同源三聚体蛋白。蛋白 S 与宿主细胞的血管紧张素转换酶 2 (ACE2) 结合,然后刺突蛋白被蛋白水解,随着病毒融合肽的暴露,蛋白构象发生剧烈变化,病毒颗粒进入宿主细胞。控制刺突蛋白构象可塑性的结构元件在很大程度上是未知的。在这里,我们提出了一种依赖于图形和中心性分析并通过生物信息学增强的方法来识别和表征蛋白质结构中的大型氢键簇。我们将这种方法应用于蛋白质 S 胞外域,发现在闭合构象中,蛋白质 S 的三个原聚体对广泛的氢键中心网络做出了相同的贡献,并且对称地对相对较大的氢键簇做出了贡献。受体结合结构域,以及靠近蛋白酶切割位点的簇。这三个簇在开放构象和融合前构象中明显不同的氢键表明动态氢键簇可以促进结构可塑性和蛋白质S原体的选择以与宿主受体结合以及蛋白水解裂解。通过对刺突蛋白序列的分析,我们确定了可能参与瞬时质子结合的组氨酸和羧酸基团的斑块。










































 京公网安备 11010802027423号
京公网安备 11010802027423号