当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Adsorption of Aromatics in MFI-Type Zeolites: Experiments and Framework Flexibility in Monte Carlo Simulations
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-09-08 , DOI: 10.1021/acs.jpcc.0c06096 Sebastián Caro-Ortiz 1 , Erik Zuidema 2 , Desmond Dekker 2 , Marcello Rigutto 2 , David Dubbeldam 3 , Thijs J. H. Vlugt 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-09-08 , DOI: 10.1021/acs.jpcc.0c06096 Sebastián Caro-Ortiz 1 , Erik Zuidema 2 , Desmond Dekker 2 , Marcello Rigutto 2 , David Dubbeldam 3 , Thijs J. H. Vlugt 1
Affiliation
|
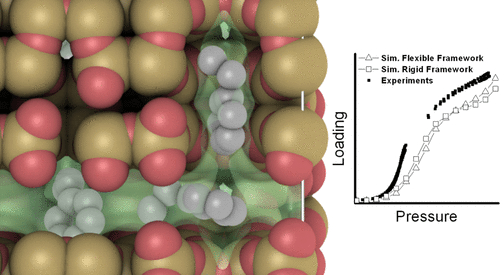
Computer simulations of adsorption of aromatics in zeolites are typically performed using rigid zeolite frameworks. However, adsorption isotherms for aromatics are very sensitive to small differences in the atomic positions of the zeolite ( Chem. Phys. Lett., 1999, 308, 155−159). This article studies the effect of framework flexibility on the adsorption of aromatics in MFI-type zeolites computed by grand-canonical Monte Carlo simulations. New experimental data of adsorption of ethylbenzene in a MFI-type zeolite at 353 K is presented. The adsorption of n-heptane, ethylbenzene, and xylene isomers is computed in three MFI-type zeolite structures. It is observed that the intraframework interactions in flexible framework models induce small but important changes in the atom positions of the zeolite and hence in the adsorption isotherms. Framework flexibility is differently “rigid”: flexible force fields produce a zeolite structure that vibrates around a new equilibrium configuration with limited capacity to accommodate to a bulky guest molecule. The vibration of the zeolite atoms only plays a role at high loadings, and the adsorption is mainly dependent on the average positions of the atoms. The simulations show that models for framework flexibility should not be blindly applied to zeolites and a general reconsideration of the parametrization schemes for such models is needed.
中文翻译:

MFI型沸石中芳烃的吸附:蒙特卡洛模拟中的实验和框架灵活性
沸石中芳烃吸附的计算机模拟通常是使用刚性沸石骨架进行的。然而,对于芳族化合物吸附等温线是在沸石(的原子位置的小差异非常敏感化学物理通讯。,1999年,308,155-159)。本文研究了通过大经典蒙特卡洛模拟计算得出的骨架柔性对MFI型沸石中芳烃吸附的影响。给出了353 K下MFI型沸石中乙苯吸附的新实验数据。n的吸附-庚烷,乙苯和二甲苯异构体以三种MFI型沸石结构计算。观察到,在柔性框架模型中的框架内相互作用引起沸石原子位置的微小但重要的变化,从而引起吸附等温线的变化。框架的柔韧性是不同的“刚性”:柔韧性场会产生一种沸石结构,该结构围绕新的平衡构型振动,而适应容纳庞大的客体分子的能力有限。沸石原子的振动仅在高载荷下起作用,吸附主要取决于原子的平均位置。仿真表明,不应将用于框架灵活性的模型盲目地应用于沸石,并且需要对此类模型的参数化方案进行一般性的重新考虑。
更新日期:2020-10-02
中文翻译:
MFI型沸石中芳烃的吸附:蒙特卡洛模拟中的实验和框架灵活性
沸石中芳烃吸附的计算机模拟通常是使用刚性沸石骨架进行的。然而,对于芳族化合物吸附等温线是在沸石(的原子位置的小差异非常敏感化学物理通讯。,1999年,308,155-159)。本文研究了通过大经典蒙特卡洛模拟计算得出的骨架柔性对MFI型沸石中芳烃吸附的影响。给出了353 K下MFI型沸石中乙苯吸附的新实验数据。n的吸附-庚烷,乙苯和二甲苯异构体以三种MFI型沸石结构计算。观察到,在柔性框架模型中的框架内相互作用引起沸石原子位置的微小但重要的变化,从而引起吸附等温线的变化。框架的柔韧性是不同的“刚性”:柔韧性场会产生一种沸石结构,该结构围绕新的平衡构型振动,而适应容纳庞大的客体分子的能力有限。沸石原子的振动仅在高载荷下起作用,吸附主要取决于原子的平均位置。仿真表明,不应将用于框架灵活性的模型盲目地应用于沸石,并且需要对此类模型的参数化方案进行一般性的重新考虑。










































 京公网安备 11010802027423号
京公网安备 11010802027423号