当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Adsorption and Dissociation of Ni(acac)2 on Iron by Ab Initio Calculations.
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-09-03 , DOI: 10.1021/acs.jpca.0c05040 Chiara Corsini 1 , Stefan Peeters 1, 2 , M C Righi 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-09-03 , DOI: 10.1021/acs.jpca.0c05040 Chiara Corsini 1 , Stefan Peeters 1, 2 , M C Righi 1
Affiliation
|
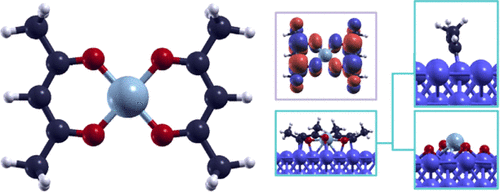
Among metal β-diketonates, nickel acetylacetonate (Ni(acac)2) has been widely employed as a precursor for many chemical structures, due to its catalytic properties. Here, we investigate, by means of density functional theory (DFT) calculations, the adsorption and dissociation of this complex: after an evaluation of the structural and electronic properties of Ni(acac)2, a comparison between different dissociation patterns reveals that the most favorable pattern for the complex adsorbed on iron is different from the one suggested by considering the strength of the bonds in the isolated complex and an attempt to generalize this dissociation model is made in this work. Moreover, the most favorable adsorption configurations turned out to be a long bridge positioning of the nickel atom along with an on top positioning of the oxygen atoms of Ni(acac)2, while a short bridge positioning is the most favorable for the central metallic unit alone.
中文翻译:

从头算计算Ni(acac)2在铁上的吸附和解离。
在金属β-二酮酸盐中,乙酰丙酮镍(Ni(acac)2)由于其催化性能而被广泛用作许多化学结构的前体。在这里,我们通过密度泛函理论(DFT)计算来研究该络合物的吸附和离解:在评估Ni(acac)2的结构和电子性质后,不同解离模式之间的比较显示出吸附在铁上的络合物的一种有利模式不同于通过考虑分离的络合物中的键的强度所建议的一种模式,并且在这项工作中尝试对这种解离模型进行泛化。而且,最有利的吸附方式竟然是长桥镍原子的定位以及Ni(acac)2的氧原子的顶部定位,而短电桥定位仅对中心金属单元最有利。
更新日期:2020-10-02
中文翻译:
从头算计算Ni(acac)2在铁上的吸附和解离。
在金属β-二酮酸盐中,乙酰丙酮镍(Ni(acac)2)由于其催化性能而被广泛用作许多化学结构的前体。在这里,我们通过密度泛函理论(DFT)计算来研究该络合物的吸附和离解:在评估Ni(acac)2的结构和电子性质后,不同解离模式之间的比较显示出吸附在铁上的络合物的一种有利模式不同于通过考虑分离的络合物中的键的强度所建议的一种模式,并且在这项工作中尝试对这种解离模型进行泛化。而且,最有利的吸附方式竟然是长桥镍原子的定位以及Ni(acac)2的氧原子的顶部定位,而短电桥定位仅对中心金属单元最有利。










































 京公网安备 11010802027423号
京公网安备 11010802027423号