当前位置:
X-MOL 学术
›
Mol. Pharmaceutics
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Hierarchical Mass Transfer Analysis of Drug Particle Dissolution, Highlighting the Hydrodynamics, pH, Particle Size, and Buffer Effects for the Dissolution of Ionizable and Nonionizable Drugs in a Compendial Dissolution Vessel.
Molecular Pharmaceutics ( IF 4.5 ) Pub Date : 2020-09-04 , DOI: 10.1021/acs.molpharmaceut.0c00614 Niloufar Salehi 1, 2 , Jozef Al-Gousous 3 , Deanna M Mudie 4 , Gordon L Amidon 2 , Robert M Ziff 1 , Gregory E Amidon 2
Molecular Pharmaceutics ( IF 4.5 ) Pub Date : 2020-09-04 , DOI: 10.1021/acs.molpharmaceut.0c00614 Niloufar Salehi 1, 2 , Jozef Al-Gousous 3 , Deanna M Mudie 4 , Gordon L Amidon 2 , Robert M Ziff 1 , Gregory E Amidon 2
Affiliation
|
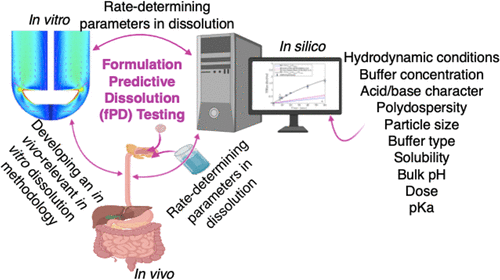
Dissolution is a crucial process for the oral delivery of drug products. Before being absorbed through epithelial cell membranes to reach the systemic circulation, drugs must first dissolve in the human gastrointestinal (GI) tract. In vivo and in vitro dissolutions are complex because of their dependency upon the drug physicochemical properties, drug product, and GI physiological properties. However, an understanding of this process is critical for the development of robust drug products. To enhance our understanding of in vivo and in vitro dissolutions, a hierarchical mass transfer (HMT) model was developed that considers the drug properties, GI fluid properties, and fluid hydrodynamics. The key drug properties include intrinsic solubility, acid/base character, pKa, particle size, and particle polydispersity. The GI fluid properties include bulk pH, buffer species concentration, fluid shear rate, and fluid convection. To corroborate the model, in vitro dissolution experiments were conducted in the United States Pharmacopeia (USP) 2 dissolution apparatus. A weakly acidic (ibuprofen), a weakly basic (haloperidol), and a nonionizable (felodipine) drug were used to study the effects of the acid/base character, pKa, and intrinsic solubility on dissolution. 900 mL of 5 mM bicarbonate and phosphate buffers at pH 6.5 and 37 °C was used to study the impact of the buffer species on drug dissolution. To investigate the impacts of fluid shear rate and convection, the apparatus was operated at different impeller rotational speeds. Moreover, presieved ibuprofen particles with different average diameters were used to investigate the effect of particle size on drug dissolution. In vitro experiments demonstrate that the dissolution rates of both the ionizable compounds used in this study were slower in bicarbonate buffer than in phosphate buffer, with the same buffer concentration, because of the lower interfacial buffer capacity, a unique behavior of bicarbonate buffer. Therefore, using surrogates (i.e., 50 mM phosphate) for bicarbonate buffer for biorelevant in vitro dissolution testing may overestimate the in vivo dissolution rate for ionizable drugs. Model simulations demonstrated that, assuming a monodisperse particle size when modeling, dissolution may overestimate the dissolution rate for polydisperse particle size distributions. The hydrodynamic parameters (maximum shear rate and fluid velocity) under in vitro conditions in the USP 2 apparatus under different rotational speeds are orders of magnitude higher compared to the in vivo situation. The inconsistencies between the in vivo and in vitro drug dissolution hydrodynamic conditions may cause an overestimation of the dissolution rate under in vitro conditions. The in vitro dissolution data supported the accuracy of the HMT for drug dissolution. This is the first drug dissolution model that incorporates the effect of the bulk pH and buffer concentration on the interfacial drug particle solubility of ionizable compounds, combined with the medium hydrodynamics effect (diffusion, convection, shear, and confinement components), and drug particle size distribution.
中文翻译:

药物颗粒溶出度的分级传质分析,重点介绍药典溶出容器中可电离和不可电离药物溶出的流体动力学、pH、粒径和缓冲作用。
溶出是药物产品口服给药的关键过程。在通过上皮细胞膜吸收到达体循环之前,药物必须首先溶解在人体胃肠 (GI) 道中。体内和体外溶出是复杂的,因为它们依赖于药物理化特性、药物产品和 GI 生理特性。然而,了解这一过程对于开发稳健的药物产品至关重要。增强我们对体内和体外的理解溶出,开发了一个分层传质 (HMT) 模型,该模型考虑了药物特性、GI 流体特性和流体流体动力学。关键的药物特性包括固有溶解度、酸/碱特性、p K a、粒径和颗粒多分散性。GI 流体特性包括体积 pH、缓冲物质浓度、流体剪切速率和流体对流。为了证实该模型,在美国药典 (USP) 2 溶出度仪中进行了体外溶出实验。弱酸性(布洛芬)、弱碱性(氟哌啶醇)和非离子化(非洛地平)药物用于研究酸/碱特性 p K a,以及溶解时的固有溶解度。使用 pH 6.5 和 37 °C 的 900 mL 5 mM 碳酸氢盐和磷酸盐缓冲液来研究缓冲液种类对药物溶出度的影响。为了研究流体剪切速率和对流的影响,该设备在不同的叶轮转速下运行。此外,使用具有不同平均直径的预筛分布洛芬颗粒来研究粒径对药物溶出度的影响。体外实验表明,本研究中使用的两种可电离化合物的溶解速率在碳酸氢盐缓冲液中比在磷酸盐缓冲液中慢,缓冲液浓度相同,因为界面缓冲能力较低,这是碳酸氢盐缓冲液的独特行为。因此,在生物相关体外溶出度测试中使用碳酸氢盐缓冲液的替代物(即 50 mM 磷酸盐)可能会高估可电离药物的体内溶出度。模型模拟表明,在建模时假设单分散粒径,溶出可能高估多分散粒径分布的溶出速率。体外流体动力学参数(最大剪切速率和流体速度)与体内情况相比,USP 2 设备在不同转速下的条件要高几个数量级。体内和体外药物溶出流体动力学条件之间的不一致可能导致体外条件下溶出速率的高估。在体外溶出度数据所支持的HMT的药物溶出的准确性。这是第一个将本体 pH 值和缓冲液浓度对可电离化合物界面药物颗粒溶解度的影响与介质流体动力学效应(扩散、对流、剪切和限制成分)和药物颗粒大小相结合的药物溶出模型分配。
更新日期:2020-10-05
中文翻译:
药物颗粒溶出度的分级传质分析,重点介绍药典溶出容器中可电离和不可电离药物溶出的流体动力学、pH、粒径和缓冲作用。
溶出是药物产品口服给药的关键过程。在通过上皮细胞膜吸收到达体循环之前,药物必须首先溶解在人体胃肠 (GI) 道中。体内和体外溶出是复杂的,因为它们依赖于药物理化特性、药物产品和 GI 生理特性。然而,了解这一过程对于开发稳健的药物产品至关重要。增强我们对体内和体外的理解溶出,开发了一个分层传质 (HMT) 模型,该模型考虑了药物特性、GI 流体特性和流体流体动力学。关键的药物特性包括固有溶解度、酸/碱特性、p K a、粒径和颗粒多分散性。GI 流体特性包括体积 pH、缓冲物质浓度、流体剪切速率和流体对流。为了证实该模型,在美国药典 (USP) 2 溶出度仪中进行了体外溶出实验。弱酸性(布洛芬)、弱碱性(氟哌啶醇)和非离子化(非洛地平)药物用于研究酸/碱特性 p K a,以及溶解时的固有溶解度。使用 pH 6.5 和 37 °C 的 900 mL 5 mM 碳酸氢盐和磷酸盐缓冲液来研究缓冲液种类对药物溶出度的影响。为了研究流体剪切速率和对流的影响,该设备在不同的叶轮转速下运行。此外,使用具有不同平均直径的预筛分布洛芬颗粒来研究粒径对药物溶出度的影响。体外实验表明,本研究中使用的两种可电离化合物的溶解速率在碳酸氢盐缓冲液中比在磷酸盐缓冲液中慢,缓冲液浓度相同,因为界面缓冲能力较低,这是碳酸氢盐缓冲液的独特行为。因此,在生物相关体外溶出度测试中使用碳酸氢盐缓冲液的替代物(即 50 mM 磷酸盐)可能会高估可电离药物的体内溶出度。模型模拟表明,在建模时假设单分散粒径,溶出可能高估多分散粒径分布的溶出速率。体外流体动力学参数(最大剪切速率和流体速度)与体内情况相比,USP 2 设备在不同转速下的条件要高几个数量级。体内和体外药物溶出流体动力学条件之间的不一致可能导致体外条件下溶出速率的高估。在体外溶出度数据所支持的HMT的药物溶出的准确性。这是第一个将本体 pH 值和缓冲液浓度对可电离化合物界面药物颗粒溶解度的影响与介质流体动力学效应(扩散、对流、剪切和限制成分)和药物颗粒大小相结合的药物溶出模型分配。










































 京公网安备 11010802027423号
京公网安备 11010802027423号