当前位置:
X-MOL 学术
›
Cryst. Growth Des.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Supramolecular Energetic and Topological Study of Halogenated Aryl Carboxylic Acids
Crystal Growth & Design ( IF 3.2 ) Pub Date : 2020-09-04 , DOI: 10.1021/acs.cgd.0c00510 Patrick Teixeira Campos 1 , Bruno Felipe Cunha da Rosa 1 , Henrique Saija Hilsinger 1 , Marcéo Auler Milani 1 , Vanessa Uecker Krüger 1
Crystal Growth & Design ( IF 3.2 ) Pub Date : 2020-09-04 , DOI: 10.1021/acs.cgd.0c00510 Patrick Teixeira Campos 1 , Bruno Felipe Cunha da Rosa 1 , Henrique Saija Hilsinger 1 , Marcéo Auler Milani 1 , Vanessa Uecker Krüger 1
Affiliation
|
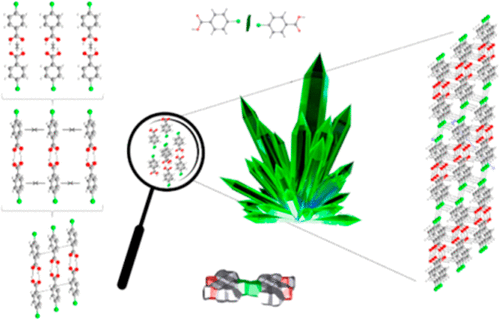
A supramolecular energetic and topological study of 12 monohalogenated aryl carboxylic acids (F, Cl, Br, I) in the ortho, meta, and para positions was performed. The energy of intermolecular interactions was determined from long-range corrected hybrid density functional theoretical computational calculations, using the ωB97X-D3 method. The surface of contact between the molecules and the molecular coordination numbers were determined. From these data, all intermolecular interactions were classified, and the robustness of the interactions was evaluated. Thus, the molecule (Mn) allocations around a central molecule (M1) are proposed. In addition, the energy from the intermolecular interactions present in the first coordination sphere, which involves the central molecule, the energy resulting from the interactions between the peripheral molecules and the total energy of the cluster provided by all interactions, was determined. The role of hydrogen bonding (O–H···O, C–H···O, and C–H···X), π···π interactions, and halogen bonding (X···X) were evaluated, in the formation of the first coordination sphere and in the nucleation process.
中文翻译:

卤代芳基羧酸的超分子能量和拓扑研究
对邻位,间位和对位的12种单卤代芳基羧酸(F,Cl,Br,I)进行了超分子能学和拓扑研究。使用ωB97X-D3方法,从远距离校正的杂化密度泛函理论计算计算中确定了分子间相互作用的能量。确定了分子之间的接触表面和分子配位数。根据这些数据,对所有分子间相互作用进行分类,并评估了相互作用的鲁棒性。因此,分子(M n)分布在中心分子(M 1)。另外,确定了存在于第一配位球中的涉及中心分子的分子间相互作用的能量,由外围分子之间的相互作用产生的能量以及由所有相互作用提供的簇的总能量。氢键(OH–·O,CH–H··O和CH–H··X),π··π相互作用和卤素键(X···X)的作用是在第一个配位球的形成和成核过程中进行了评估。
更新日期:2020-10-07
中文翻译:
卤代芳基羧酸的超分子能量和拓扑研究
对邻位,间位和对位的12种单卤代芳基羧酸(F,Cl,Br,I)进行了超分子能学和拓扑研究。使用ωB97X-D3方法,从远距离校正的杂化密度泛函理论计算计算中确定了分子间相互作用的能量。确定了分子之间的接触表面和分子配位数。根据这些数据,对所有分子间相互作用进行分类,并评估了相互作用的鲁棒性。因此,分子(M n)分布在中心分子(M 1)。另外,确定了存在于第一配位球中的涉及中心分子的分子间相互作用的能量,由外围分子之间的相互作用产生的能量以及由所有相互作用提供的簇的总能量。氢键(OH–·O,CH–H··O和CH–H··X),π··π相互作用和卤素键(X···X)的作用是在第一个配位球的形成和成核过程中进行了评估。








































 京公网安备 11010802027423号
京公网安备 11010802027423号