当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular dynamics studies on formation of stacking fault tetrahedra in FCC metals
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-01-01 , DOI: 10.1016/j.commatsci.2020.110017 Arun Kumar Panda , R. Divakar , Akash Singh , R. Thirumurugesan , P. Parameswaran
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-01-01 , DOI: 10.1016/j.commatsci.2020.110017 Arun Kumar Panda , R. Divakar , Akash Singh , R. Thirumurugesan , P. Parameswaran
|
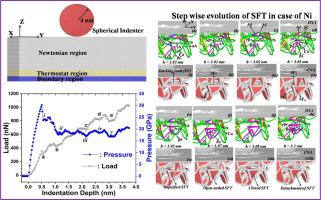
Abstract The formation of stacking fault tetrahedra (SFT) based on the vacancy clustering mechanism and Frank loop in case of face centered cubic (FCC) metals and alloys is widely accepted, but the corresponding research for SFT formation without the involvement of Frank loop and interaction of dislocations is not well understood. In this article, we attempt to explain the formation of SFT from vacancies, voids and nanoindentation process in case of low stacking fault energy (Ag) and high stacking fault energy (Ni) FCC metals. The atomic scale processes are simulated and step wise evolution of vacancy cluster and dislocation loops to SFT are provided by molecular dynamics method. We describe in this atomistic simulation that the voids containing 13 and 43 vacancy atoms can rearrange themselves to transform to SFT without going through the Frank loop. The present work has also demonstrated the formation of SFT during nano indentation of (1 1 1) surface of Ag and Ni through interactions of Shockley partials without the involvement of vacancies or Frank loop.
中文翻译:

FCC金属层错四面体形成的分子动力学研究
摘要 面心立方 (FCC) 金属和合金中基于空位聚集机制和 Frank 环的层错四面体 (SFT) 的形成被广泛接受,但相应的 SFT 形成研究并未涉及 Frank 环和相互作用对位错的理解不是很清楚。在本文中,我们试图解释在低层错能 (Ag) 和高层错能 (Ni) FCC 金属的情况下,由空位、空隙和纳米压痕过程形成 SFT。模拟原子尺度过程,并通过分子动力学方法提供空位簇和位错环到 SFT 的逐步演化。我们在这个原子模拟中描述了包含 13 和 43 个空位原子的空隙可以重新排列以转变为 SFT,而无需通过 Frank 循环。目前的工作还证明了在 Ag 和 Ni 的 (1 1 1) 表面纳米压痕过程中通过肖克利部分的相互作用形成了 SFT,而没有空位或弗兰克环的参与。
更新日期:2021-01-01
中文翻译:
FCC金属层错四面体形成的分子动力学研究
摘要 面心立方 (FCC) 金属和合金中基于空位聚集机制和 Frank 环的层错四面体 (SFT) 的形成被广泛接受,但相应的 SFT 形成研究并未涉及 Frank 环和相互作用对位错的理解不是很清楚。在本文中,我们试图解释在低层错能 (Ag) 和高层错能 (Ni) FCC 金属的情况下,由空位、空隙和纳米压痕过程形成 SFT。模拟原子尺度过程,并通过分子动力学方法提供空位簇和位错环到 SFT 的逐步演化。我们在这个原子模拟中描述了包含 13 和 43 个空位原子的空隙可以重新排列以转变为 SFT,而无需通过 Frank 循环。目前的工作还证明了在 Ag 和 Ni 的 (1 1 1) 表面纳米压痕过程中通过肖克利部分的相互作用形成了 SFT,而没有空位或弗兰克环的参与。










































 京公网安备 11010802027423号
京公网安备 11010802027423号