Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Machine learning corrected alchemical perturbation density functional theory for catalysis applications
AIChE Journal ( IF 3.5 ) Pub Date : 2020-09-02 , DOI: 10.1002/aic.17041 Charles D. Griego 1 , Lingyan Zhao 1 , Karthikeyan Saravanan 1 , John A. Keith 1
AIChE Journal ( IF 3.5 ) Pub Date : 2020-09-02 , DOI: 10.1002/aic.17041 Charles D. Griego 1 , Lingyan Zhao 1 , Karthikeyan Saravanan 1 , John A. Keith 1
Affiliation
|
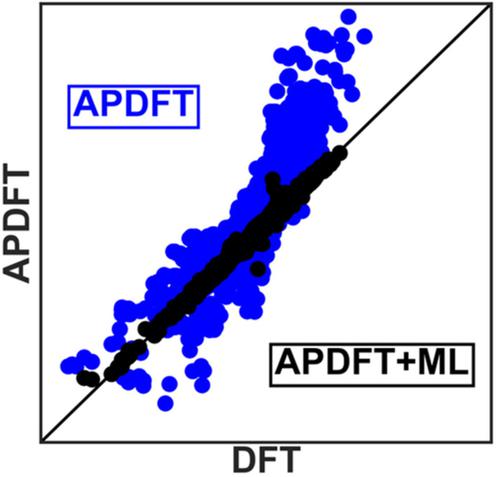
Alchemical perturbation density functional theory (APDFT) has promise for enabling computational screening of hypothetical catalyst sites. Here, we analyze errors in first order APDFT calculation schemes for binding energies of CHx, NHx, OHx, and OOH adsorbates over a range of different coverages on hypothetical alloys based on a Pt(111) reference system. We then train three different support vector regression machine learning models that correct systematic APDFT prediction errors for each of the three classes of carbon, nitrogen, and oxygen based adsorbates. While uncorrected first order APDFT alone approximates accurate adsorbate binding energies on up to 36 hypothetical alloys based on a single Kohn–Sham DFT calculation on a 3 × 3 unit cell for Pt(111), the machine learning‐corrected APDFT extends this number to more than 20,000 and provides a recipe for developing other machine learning‐based APDFT models.
中文翻译:

机器学习校正的炼金术扰动密度泛函理论在催化中的应用
炼金术扰动密度泛函理论(APDFT)有望实现对假设催化剂位点的计算筛选。在这里,我们分析一阶APDFT计算方案中CH x,NH x,OH x的结合能的误差,并且OOH在基于Pt(111)参考系统的假设合金上具有一系列不同的覆盖率。然后,我们训练三种不同的支持向量回归机器学习模型,这些模型针对碳,氮和氧基吸附物这三类中的每一种纠正系统性APDFT预测误差。根据3个3×3晶胞对Pt(111)进行的一次Kohn-Sham DFT计算,仅未经校正的一阶APDFT可以近似估计多达36种假设合金的准确吸附物结合能,而经过机器学习校正的APDFT可以将该数字扩展到更多超过20,000,并提供了开发其他基于机器学习的APDFT模型的秘诀。
更新日期:2020-09-02
中文翻译:
机器学习校正的炼金术扰动密度泛函理论在催化中的应用
炼金术扰动密度泛函理论(APDFT)有望实现对假设催化剂位点的计算筛选。在这里,我们分析一阶APDFT计算方案中CH x,NH x,OH x的结合能的误差,并且OOH在基于Pt(111)参考系统的假设合金上具有一系列不同的覆盖率。然后,我们训练三种不同的支持向量回归机器学习模型,这些模型针对碳,氮和氧基吸附物这三类中的每一种纠正系统性APDFT预测误差。根据3个3×3晶胞对Pt(111)进行的一次Kohn-Sham DFT计算,仅未经校正的一阶APDFT可以近似估计多达36种假设合金的准确吸附物结合能,而经过机器学习校正的APDFT可以将该数字扩展到更多超过20,000,并提供了开发其他基于机器学习的APDFT模型的秘诀。










































 京公网安备 11010802027423号
京公网安备 11010802027423号