当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Constrained Density Functional Theory Molecular Dynamics Simulation of Deprotonation in Aqueous Silicic Acid.
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2020-08-28 , DOI: 10.1021/acs.jpcb.0c05096 Tatsuya Joutsuka 1 , Koji Ando 2
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2020-08-28 , DOI: 10.1021/acs.jpcb.0c05096 Tatsuya Joutsuka 1 , Koji Ando 2
Affiliation
|
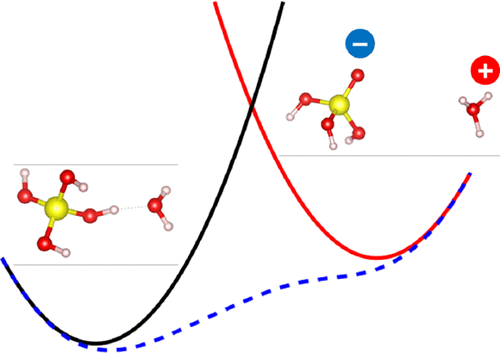
The solubility of silicic acid in water is so low that the molecular mechanism behind the physical properties such as pKa remains poorly understood, despite the importance in fields such as chemistry and geology. Theoretical calculations provide the molecular-level information on such a rare chemical species yet face difficulties in the selection of reaction coordinates and the rare-event sampling of proton transfers (PTs) in condensed phases. We propose a straightforward calculation scheme of pKa with ab initio molecular dynamics (MD) simulation and the constrained density functional theory (CDFT), which provides structural and dynamical properties such as radial distribution functions, vibrational spectra, and reaction paths. The calculated reaction free energies of deprotonations agreed with experiments within a few kcal/mol. Analysis of the solvation structure shows that, after deprotonation, the hydronium ion D3O+ repels away from the deprotonated silicic acid SiO(OD)3– without forming a stable contact-ion pair. The calculated vibrational spectra are consistent with the spectroscopic measurements, and the dynamical analysis of the reaction path quantifies the couplings of the OD stretch vibrations of silicic acid and water to the PT reaction in terms of the vertical energy-gap coordinate that includes both the solute and the solvent components.
中文翻译:

约束密度泛函理论在硅酸水溶液中去质子化的分子动力学模拟。
硅酸在水中的溶解度如此之低,尽管在诸如化学和地质学等领域具有重要意义,但对诸如p K a的物理性质背后的分子机理仍然知之甚少。理论计算提供了有关这种稀有化学物质的分子水平信息,但在选择反应坐标和凝聚相中质子转移(PTs)的稀有事件采样时面临困难。我们建议p的简单计算方案ķ一个与从头分子动力学(MD)模拟和约束密度泛函理论(CDFT),它提供了结构和动力学特性,例如径向分布函数,振动谱和反应路径。计算的去质子反应自由能与实验在几千卡/摩尔之内一致。对溶剂化结构的分析表明,去质子化后,水合氢离子D 3 O +排斥已去质子化的硅酸SiO(OD)3 –没有形成稳定的接触离子对。计算出的振动光谱与光谱测量结果一致,反应路径的动力学分析根据包括两个溶质在内的垂直能隙坐标,量化了硅酸和水的OD拉伸振动与PT反应的耦合。和溶剂成分。
更新日期:2020-09-24
中文翻译:
约束密度泛函理论在硅酸水溶液中去质子化的分子动力学模拟。
硅酸在水中的溶解度如此之低,尽管在诸如化学和地质学等领域具有重要意义,但对诸如p K a的物理性质背后的分子机理仍然知之甚少。理论计算提供了有关这种稀有化学物质的分子水平信息,但在选择反应坐标和凝聚相中质子转移(PTs)的稀有事件采样时面临困难。我们建议p的简单计算方案ķ一个与从头分子动力学(MD)模拟和约束密度泛函理论(CDFT),它提供了结构和动力学特性,例如径向分布函数,振动谱和反应路径。计算的去质子反应自由能与实验在几千卡/摩尔之内一致。对溶剂化结构的分析表明,去质子化后,水合氢离子D 3 O +排斥已去质子化的硅酸SiO(OD)3 –没有形成稳定的接触离子对。计算出的振动光谱与光谱测量结果一致,反应路径的动力学分析根据包括两个溶质在内的垂直能隙坐标,量化了硅酸和水的OD拉伸振动与PT反应的耦合。和溶剂成分。










































 京公网安备 11010802027423号
京公网安备 11010802027423号