当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
On the X-ray Scattering Pre-peak of Linear Mono-ols and the Related Microstructure from Computer Simulations.
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2020-08-28 , DOI: 10.1021/acs.jpcb.0c05932 Martina Požar 1 , Jennifer Bolle 2 , Christian Sternemann 2 , Aurélien Perera 3
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2020-08-28 , DOI: 10.1021/acs.jpcb.0c05932 Martina Požar 1 , Jennifer Bolle 2 , Christian Sternemann 2 , Aurélien Perera 3
Affiliation
|
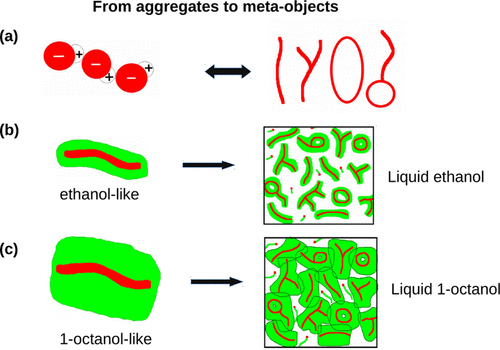
The X-ray scattering intensities (I(k)) of linear alkanols OH(CH2)n−1CH3 obtained from experiments (methanol to 1-undecanol) and computer simulations (methanol to 1-nonanol) of different force field models are comparatively studied particularly in order to explain the origin and the properties of the scattering pre-peak in the k-vector range 0.3–1 Å–1. The experimental I(k) values show two apparent features: the pre-peak position kP decreases with increasing n, and more intriguingly, the amplitude AP goes through a maximum at 1-butanol (n = 4). The first feature is well reproduced by all force-field models, while the second shows strong model dependence. The simulations reveal various shapes of clusters of the hydroxyl head-group from n>2. kP is directly related to the size of the meta-objects corresponding to such clusters surrounded by their alkyl tails. The explanation of the AP turnover at n = 4 is more involved in terms of cancellations of atom–atom structure factor S(k) contributions related to domain ordering. The flexibility of the alkyl tails tends to reduce the cross contributions, thus revealing the crucial importance of this parameter in the models. Force fields with all-atom representation are less successful in reproducing the pre-peak features for smaller alkanols, n<6, possibly because they blur the charge ordering process since all atoms bear partial charges. The analysis clearly shows that it is not possible to obtain a model-free explanation of the features of I(k).
中文翻译:

线性单醇的X射线散射峰峰及相关的微观结构的计算机模拟。
线性链烷醇OH(CH 2)n -1 CH 3的X射线散射强度(I(k))从不同力场模型的实验(甲醇到1-十一烷醇)和计算机模拟(甲醇到1-壬醇)获得我们进行了比较研究,目的是为了解释k矢量范围0.3–1 Å –1中散射峰的起源和性质。实验I(k)值表现出两个明显的特征:峰前位置k P随着n的增加而减小,并且更有趣的是,振幅A P在1-丁醇处经历最大值(n = 4)。所有力场模型都很好地再现了第一个特征,而第二个特征则显示出强烈的模型依赖性。模拟揭示了n > 2时羟基头基团簇的各种形状。k P与与被其烷基尾部包围的簇相对应的元对象的大小直接相关。关于n = 4时A P转换的解释更多地涉及原子-原子结构因子S(k)与域排序相关的贡献。烷基尾巴的柔韧性往往会降低交叉贡献,因此揭示了该参数在模型中的至关重要性。具有全原子表示的力场在再现较小的n <6的较小链烷醇的峰前特征方面不太成功,这可能是因为它们模糊了电荷排序过程,因为所有原子都带有部分电荷。分析清楚地表明,不可能获得I(k)特征的无模型解释。
更新日期:2020-09-24
中文翻译:
线性单醇的X射线散射峰峰及相关的微观结构的计算机模拟。
线性链烷醇OH(CH 2)n -1 CH 3的X射线散射强度(I(k))从不同力场模型的实验(甲醇到1-十一烷醇)和计算机模拟(甲醇到1-壬醇)获得我们进行了比较研究,目的是为了解释k矢量范围0.3–1 Å –1中散射峰的起源和性质。实验I(k)值表现出两个明显的特征:峰前位置k P随着n的增加而减小,并且更有趣的是,振幅A P在1-丁醇处经历最大值(n = 4)。所有力场模型都很好地再现了第一个特征,而第二个特征则显示出强烈的模型依赖性。模拟揭示了n > 2时羟基头基团簇的各种形状。k P与与被其烷基尾部包围的簇相对应的元对象的大小直接相关。关于n = 4时A P转换的解释更多地涉及原子-原子结构因子S(k)与域排序相关的贡献。烷基尾巴的柔韧性往往会降低交叉贡献,因此揭示了该参数在模型中的至关重要性。具有全原子表示的力场在再现较小的n <6的较小链烷醇的峰前特征方面不太成功,这可能是因为它们模糊了电荷排序过程,因为所有原子都带有部分电荷。分析清楚地表明,不可能获得I(k)特征的无模型解释。










































 京公网安备 11010802027423号
京公网安备 11010802027423号