当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Fluorination Position: A Study of the Optoelectronic Properties of Two Regioisomers Using Spectroscopic and Computational Techniques.
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-08-28 , DOI: 10.1021/acs.jpca.0c05210 Joshua J Sutton 1 , Yuxiang Li 2 , Hwa Sook Ryu 2 , Elliot J Tay 1 , Han Young Woo 2 , Keith C Gordon 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-08-28 , DOI: 10.1021/acs.jpca.0c05210 Joshua J Sutton 1 , Yuxiang Li 2 , Hwa Sook Ryu 2 , Elliot J Tay 1 , Han Young Woo 2 , Keith C Gordon 1
Affiliation
|
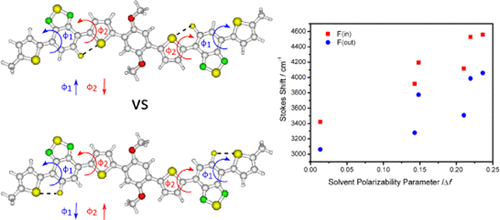
The influence of the fluorination positions on the optical and electronic properties of a pair of donor–acceptor (DA)-based regioisomers is explored. In the regioisomers, the fluorination was varied between the 2 and 3 positions on benzothiadiazole (BTD) acceptor units. Although the structural variation between the regioisomers is small, significant variations in the electronic properties of the two compounds were observed. These were observed most markedly in the excited-state properties with a 15 nm (280–390 cm–1) red shift of the emission between the regioisomers. The combination of resonance Raman spectroscopy (RRS) and density functional theory (DFT) calculations was used to probe the possible causes of the observed variations. The analysis suggested a F···S through-space interaction as being responsible for tuning both the electronic properties and rigidity of the compounds. Shifting the fluorine atoms shifted the location of the F···S interaction, changing which part of the molecule was locked down, and showed a variation in the overall rigidity of the molecule. In this series, the influence could be varied between the core and periphery. This study adds to a growing body of work demonstrating the effectiveness of selective fluorination in tailoring the properties of organic molecules.
中文翻译:

氟化位置:使用光谱和计算技术研究两种区域异构体的光电性质。
探索了氟化位置对一对基于供体-受体(DA)的区域异构体的光学和电子性质的影响。在区域异构体中,氟在苯并噻二唑(BTD)受体单元的2和3位之间变化。尽管区域异构体之间的结构变化很小,但是观察到两种化合物的电子性质的显着变化。在15 nm(280–390 cm –1)的激发态性质中,这些现象最为明显。)区域异构体之间的发射红移。共振拉曼光谱法(RRS)和密度泛函理论(DFT)计算的结合被用来探测观察到的变化的可能原因。分析表明,F···S贯穿空间相互作用是调节化合物的电子性能和刚性的原因。氟原子的移动改变了F···S相互作用的位置,改变了分子的哪个部分被锁定,并显示了分子整体刚度的变化。在这个系列中,影响可能在核心和外围之间变化。这项研究增加了越来越多的工作量,证明了选择性氟化在调整有机分子特性方面的有效性。
更新日期:2020-09-24
中文翻译:
氟化位置:使用光谱和计算技术研究两种区域异构体的光电性质。
探索了氟化位置对一对基于供体-受体(DA)的区域异构体的光学和电子性质的影响。在区域异构体中,氟在苯并噻二唑(BTD)受体单元的2和3位之间变化。尽管区域异构体之间的结构变化很小,但是观察到两种化合物的电子性质的显着变化。在15 nm(280–390 cm –1)的激发态性质中,这些现象最为明显。)区域异构体之间的发射红移。共振拉曼光谱法(RRS)和密度泛函理论(DFT)计算的结合被用来探测观察到的变化的可能原因。分析表明,F···S贯穿空间相互作用是调节化合物的电子性能和刚性的原因。氟原子的移动改变了F···S相互作用的位置,改变了分子的哪个部分被锁定,并显示了分子整体刚度的变化。在这个系列中,影响可能在核心和外围之间变化。这项研究增加了越来越多的工作量,证明了选择性氟化在调整有机分子特性方面的有效性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号