当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Assessment of Force Field Accuracy Using Cryogenic Electron Microscopy Data of Hyper-thermostable Glutamate Dehydrogenase.
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2020-08-25 , DOI: 10.1021/acs.jpcb.0c04464 Tomotaka Oroguchi 1, 2 , Mao Oide 1, 2 , Taiki Wakabayashi 1, 2 , Masayoshi Nakasako 1, 2
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2020-08-25 , DOI: 10.1021/acs.jpcb.0c04464 Tomotaka Oroguchi 1, 2 , Mao Oide 1, 2 , Taiki Wakabayashi 1, 2 , Masayoshi Nakasako 1, 2
Affiliation
|
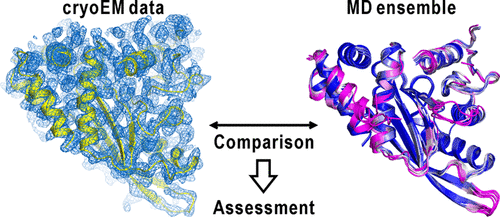
Molecular dynamics (MD) simulations in biophysically relevant time scales of microseconds is a powerful tool for studying biomolecular processes, but results often display force field dependency. Therefore, assessment of force field accuracy using experimental data of biomolecules in solution is essential for simulation studies. Here, we propose the use of structural models obtained via cryo-electron microscopy (cryoEM), which provides biomolecular structures in vitreous ice mimicking the environment in solution. The accuracy of the AMBER (ff99SB-ILDN-NMR, ff14SB, ff15ipq, and ff15FB) and CHARMM (CHARMM22 and CHARMM36m) force fields was assessed by comparing their MD trajectories with the cryoEM data of thermostable hexameric glutamate dehydrogenase (GDH), which included a cryoEM map at a resolution of approximately 3 Å and structure models of subunits reflecting metastable conformations in domain motion occurring in GDH. In the assessment, we validated the force fields with respect to the reproducibility and stability of secondary structures and intersubunit interactions in the cryoEM data. Furthermore, we evaluated the force fields regarding the reproducibility of the energy landscape in the domain motion expected from the cryoEM data. As a result, among the six force fields, ff15FB and ff99SB-ILDN-NMR displayed good agreement with the experiment. The present study demonstrated the advantages of the high-resolution cryoEM map and suggested the optimal force field to reproduce experimentally observed protein structures.
中文翻译:

使用超耐热谷氨酸脱氢酶的低温电子显微镜数据评估力场精度。
在生物物理相关的微秒时间尺度上的分子动力学(MD)模拟是研究生物分子过程的强大工具,但结果通常显示出力场依赖性。因此,使用溶液中生物分子的实验数据评估力场精度对于仿真研究至关重要。在这里,我们建议使用通过低温电子显微镜(cryoEM)获得的结构模型,该模型在玻璃冰中提供了模拟溶液中环境的生物分子结构。通过将它们的MD轨迹与热稳定的六聚谷氨酸脱氢酶(GDH)的cryoEM数据进行比较,来评估AMBER(ff99SB-ILDN-NMR,ff14SB,ff15ipq和ff15FB)和CHARMM(CHARMM22和CHARMM36m)力场的准确性 其中包括分辨率约为3Å的cryoEM图和反映GDH中域运动中亚稳态构象的亚基结构模型。在评估中,我们验证了cryoEM数据中有关二级结构的可再现性和稳定性以及亚基间相互作用的力场。此外,我们评估了根据cryoEM数据预期的域运动中能量景观可重复性的力场。结果,在六个力场中,ff15FB和ff99SB-ILDN-NMR与实验显示出良好的一致性。本研究证明了高分辨率cryoEM图的优势,并建议了最佳力场来重现实验观察到的蛋白质结构。在评估中,我们验证了cryoEM数据中有关二级结构的可再现性和稳定性以及亚基间相互作用的力场。此外,我们评估了根据cryoEM数据预期的域运动中能量景观可重复性的力场。结果,在六个力场中,ff15FB和ff99SB-ILDN-NMR与实验显示出良好的一致性。本研究证明了高分辨率cryoEM图的优势,并建议了最佳力场来重现实验观察到的蛋白质结构。在评估中,我们验证了cryoEM数据中有关二级结构的可再现性和稳定性以及亚基间相互作用的力场。此外,我们评估了根据cryoEM数据预期的域运动中能量景观可重复性的力场。结果,在六个力场中,ff15FB和ff99SB-ILDN-NMR与实验显示出良好的一致性。本研究证明了高分辨率cryoEM图的优势,并建议了最佳力场来重现实验观察到的蛋白质结构。我们评估了与cryoEM数据预期的域运动中能量分布的可重复性有关的力场。结果,在六个力场中,ff15FB和ff99SB-ILDN-NMR与实验显示出良好的一致性。本研究证明了高分辨率cryoEM图的优势,并建议了最佳力场来重现实验观察到的蛋白质结构。我们评估了与cryoEM数据预期的域运动中能量分布的可重复性有关的力场。结果,在六个力场中,ff15FB和ff99SB-ILDN-NMR与实验显示出良好的一致性。本研究证明了高分辨率cryoEM图的优势,并建议了最佳力场来重现实验观察到的蛋白质结构。
更新日期:2020-10-02
中文翻译:
使用超耐热谷氨酸脱氢酶的低温电子显微镜数据评估力场精度。
在生物物理相关的微秒时间尺度上的分子动力学(MD)模拟是研究生物分子过程的强大工具,但结果通常显示出力场依赖性。因此,使用溶液中生物分子的实验数据评估力场精度对于仿真研究至关重要。在这里,我们建议使用通过低温电子显微镜(cryoEM)获得的结构模型,该模型在玻璃冰中提供了模拟溶液中环境的生物分子结构。通过将它们的MD轨迹与热稳定的六聚谷氨酸脱氢酶(GDH)的cryoEM数据进行比较,来评估AMBER(ff99SB-ILDN-NMR,ff14SB,ff15ipq和ff15FB)和CHARMM(CHARMM22和CHARMM36m)力场的准确性 其中包括分辨率约为3Å的cryoEM图和反映GDH中域运动中亚稳态构象的亚基结构模型。在评估中,我们验证了cryoEM数据中有关二级结构的可再现性和稳定性以及亚基间相互作用的力场。此外,我们评估了根据cryoEM数据预期的域运动中能量景观可重复性的力场。结果,在六个力场中,ff15FB和ff99SB-ILDN-NMR与实验显示出良好的一致性。本研究证明了高分辨率cryoEM图的优势,并建议了最佳力场来重现实验观察到的蛋白质结构。在评估中,我们验证了cryoEM数据中有关二级结构的可再现性和稳定性以及亚基间相互作用的力场。此外,我们评估了根据cryoEM数据预期的域运动中能量景观可重复性的力场。结果,在六个力场中,ff15FB和ff99SB-ILDN-NMR与实验显示出良好的一致性。本研究证明了高分辨率cryoEM图的优势,并建议了最佳力场来重现实验观察到的蛋白质结构。在评估中,我们验证了cryoEM数据中有关二级结构的可再现性和稳定性以及亚基间相互作用的力场。此外,我们评估了根据cryoEM数据预期的域运动中能量景观可重复性的力场。结果,在六个力场中,ff15FB和ff99SB-ILDN-NMR与实验显示出良好的一致性。本研究证明了高分辨率cryoEM图的优势,并建议了最佳力场来重现实验观察到的蛋白质结构。我们评估了与cryoEM数据预期的域运动中能量分布的可重复性有关的力场。结果,在六个力场中,ff15FB和ff99SB-ILDN-NMR与实验显示出良好的一致性。本研究证明了高分辨率cryoEM图的优势,并建议了最佳力场来重现实验观察到的蛋白质结构。我们评估了与cryoEM数据预期的域运动中能量分布的可重复性有关的力场。结果,在六个力场中,ff15FB和ff99SB-ILDN-NMR与实验显示出良好的一致性。本研究证明了高分辨率cryoEM图的优势,并建议了最佳力场来重现实验观察到的蛋白质结构。










































 京公网安备 11010802027423号
京公网安备 11010802027423号