当前位置:
X-MOL 学术
›
J. Org. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Strategy Used to Control the Mechanism of Homogeneous Alkyne/Olefin Hydrogenation: AIMD Simulations and DFT Calculations.
The Journal of Organic Chemistry ( IF 3.3 ) Pub Date : 2020-08-24 , DOI: 10.1021/acs.joc.0c01021 Yafei Luo 1 , Zheng Huang 1 , Zhongzhu Chen 1 , Zhigang Xu 1 , Jiangping Meng 1 , Hong-Yu Li 2 , Qingxi Meng 3 , Dianyong Tang 1
The Journal of Organic Chemistry ( IF 3.3 ) Pub Date : 2020-08-24 , DOI: 10.1021/acs.joc.0c01021 Yafei Luo 1 , Zheng Huang 1 , Zhongzhu Chen 1 , Zhigang Xu 1 , Jiangping Meng 1 , Hong-Yu Li 2 , Qingxi Meng 3 , Dianyong Tang 1
Affiliation
|
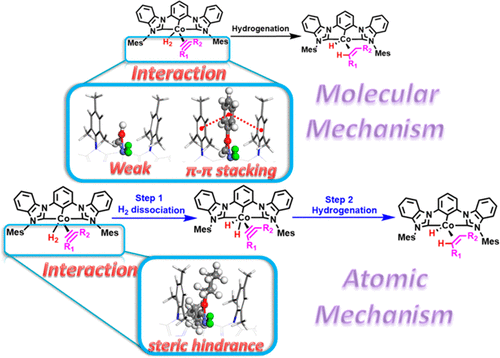
Understanding the mechanism of the catalytic reaction is an effective way to design new high-performance catalysts. The mechanisms of alkyne/olefin hydrogenations catalyzed by a nonclassical Co–N2 catalyst are explored by ab initio molecular dynamics simulations and density functional theory calculations. From the calculated results, the hydrogenation mechanisms, i.e., molecular or atomic mechanisms, can be effectively controlled via employing the different interaction between the catalyst and substrates. The origination of excellent selectivity toward E-olefins for the Co–N2 catalyst is also taken into account with the help of investigating the olefin hydrogenation process. The mechanism indicates that the negligible energy barrier of rotation is the main reason for highly selective semihydrogenation of a Co–N2 catalyst, which leads to the trans-olefin formation. These investigations may provide some useful information and guidelines on the current understanding of the hydrogenation reaction and designing the high-performance catalysts.
中文翻译:

控制均相炔烃/烯烃加氢机理的策略:AIMD模拟和DFT计算。
了解催化反应的机理是设计新型高性能催化剂的有效途径。通过从头算分子动力学模拟和密度泛函理论计算,探索了非经典Co–N 2催化剂催化的炔烃/烯烃加氢机理。根据计算结果,可以通过利用催化剂和底物之间的不同相互作用来有效地控制氢化机理,即分子或原子机理。Co-N 2对E-烯烃具有优异的选择性的起源在研究烯烃氢化过程的帮助下,还考虑了催化剂。该机理表明,可忽略的旋转能垒是Co-N 2催化剂高度选择性半氢化反应的主要原因,这导致反式烯烃的形成。这些研究可能会提供一些有用的信息和指南,以帮助您了解当前的加氢反应和设计高性能催化剂。
更新日期:2020-09-20
中文翻译:
控制均相炔烃/烯烃加氢机理的策略:AIMD模拟和DFT计算。
了解催化反应的机理是设计新型高性能催化剂的有效途径。通过从头算分子动力学模拟和密度泛函理论计算,探索了非经典Co–N 2催化剂催化的炔烃/烯烃加氢机理。根据计算结果,可以通过利用催化剂和底物之间的不同相互作用来有效地控制氢化机理,即分子或原子机理。Co-N 2对E-烯烃具有优异的选择性的起源在研究烯烃氢化过程的帮助下,还考虑了催化剂。该机理表明,可忽略的旋转能垒是Co-N 2催化剂高度选择性半氢化反应的主要原因,这导致反式烯烃的形成。这些研究可能会提供一些有用的信息和指南,以帮助您了解当前的加氢反应和设计高性能催化剂。










































 京公网安备 11010802027423号
京公网安备 11010802027423号