当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Conformational ensembles of an intrinsically disordered protein consistent with NMR, SAXS and single-molecule FRET
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2020-08-25 , DOI: 10.1021/jacs.0c02088 Gregory-Neal W Gomes 1, 2 , Mickaël Krzeminski 3, 4 , Ashley Namini 1, 2 , Erik W Martin 5 , Tanja Mittag 5 , Teresa Head-Gordon 6 , Julie D Forman-Kay 3, 4 , Claudiu C Gradinaru 1, 2
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2020-08-25 , DOI: 10.1021/jacs.0c02088 Gregory-Neal W Gomes 1, 2 , Mickaël Krzeminski 3, 4 , Ashley Namini 1, 2 , Erik W Martin 5 , Tanja Mittag 5 , Teresa Head-Gordon 6 , Julie D Forman-Kay 3, 4 , Claudiu C Gradinaru 1, 2
Affiliation
|
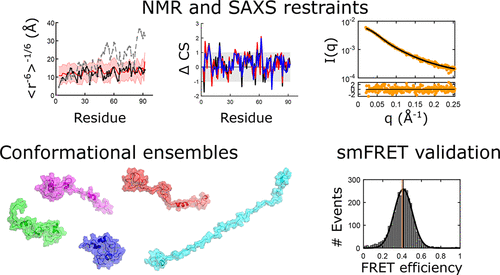
Intrinsically disordered proteins (IDPs) have fluctuating heterogeneous conformations, which makes structural characterization challenging, but of great interest, since their conformational ensembles are the link between their sequences and functions. An accurate description of IDP conformational ensembles depends crucially on the amount and quality of the experimental data, how it is integrated, and if it mutually supports a consistent structural picture. We used integrative modelling and validation to apply conformational restraints and assess agreement with the most common structural techniques for IDPs: Nuclear Magnetic Resonance (NMR) spectroscopy, Small-angle X-ray Scattering (SAXS), and single-molecule Förster Resonance Energy Transfer (smFRET) reach concordance on structural ensembles for Sic1 and phosphorylated Sic1 (pSic1). To resolve apparent discrepancies between smFRET and SAXS, we integrated SAXS data with non-smFRET (NMR) data and reserved the new smFRET data for Sic1 and pSic1, as an independent validation. Given the consistency of the SAXS/PRE restrained ensembles with smFRET, which was not guaranteed a priori, indicates that the perturbative effects of NMR or smFRET labels on the Sic1 and pSic1 ensemble are minimal. Furthermore , the mutual agreement with such a diverse set of experimental data suggests that details of the generated ensembles can now be examined with a high degree of confidence, such as overall compactness and end-to-end distance fluctuations, to reveal distinguishing features of Sic1 vs. pSic1. From the experimentally well supported ensembles, we find they are consistent with independent biophysical models of Sic1's ultrasensitive binding to its partner Cdc4. Our results underscore the importance of integrative modelling and validation in calculating and drawing biological conclusions from IDP conformational ensembles.
中文翻译:

与 NMR、SAXS 和单分子 FRET 一致的本质无序蛋白质的构象集合
本质上无序的蛋白质 (IDP) 具有波动的异质构象,这使得结构表征具有挑战性,但非常有趣,因为它们的构象集合是它们的序列和功能之间的联系。IDP 构象集合的准确描述关键取决于实验数据的数量和质量、它是如何整合的,以及它是否相互支持一致的结构图。我们使用集成建模和验证来应用构象限制并评估与 IDP 最常见结构技术的一致性:核磁共振 (NMR) 光谱、小角 X 射线散射 (SAXS) 和单分子 Förster 共振能量转移 ( smFRET) 在 Sic1 和磷酸化 Sic1 (pSic1) 的结构整体上达成一致性。为了解决 smFRET 和 SAXS 之间的明显差异,我们将 SAXS 数据与非 smFRET (NMR) 数据整合在一起,并为 Sic1 和 pSic1 保留了新的 smFRET 数据,作为独立验证。鉴于 SAXS/PRE 约束合奏与 smFRET 的一致性,这不是先验保证,表明 NMR 或 smFRET 标签对 Sic1 和 pSic1 合奏的微扰影响很小。此外,与如此多样化的实验数据集的相互一致表明,现在可以高度自信地检查生成的集合的细节,例如整体紧凑性和端到端距离波动,以揭示 Sic1 的显着特征与 pSic1 相比。从实验得到很好支持的合奏中,我们发现它们与 Sic1' 的独立生物物理模型一致 s 与其合作伙伴 Cdc4 的超灵敏结合。我们的结果强调了综合建模和验证在从 IDP 构象集合计算和得出生物学结论中的重要性。
更新日期:2020-08-25
中文翻译:
与 NMR、SAXS 和单分子 FRET 一致的本质无序蛋白质的构象集合
本质上无序的蛋白质 (IDP) 具有波动的异质构象,这使得结构表征具有挑战性,但非常有趣,因为它们的构象集合是它们的序列和功能之间的联系。IDP 构象集合的准确描述关键取决于实验数据的数量和质量、它是如何整合的,以及它是否相互支持一致的结构图。我们使用集成建模和验证来应用构象限制并评估与 IDP 最常见结构技术的一致性:核磁共振 (NMR) 光谱、小角 X 射线散射 (SAXS) 和单分子 Förster 共振能量转移 ( smFRET) 在 Sic1 和磷酸化 Sic1 (pSic1) 的结构整体上达成一致性。为了解决 smFRET 和 SAXS 之间的明显差异,我们将 SAXS 数据与非 smFRET (NMR) 数据整合在一起,并为 Sic1 和 pSic1 保留了新的 smFRET 数据,作为独立验证。鉴于 SAXS/PRE 约束合奏与 smFRET 的一致性,这不是先验保证,表明 NMR 或 smFRET 标签对 Sic1 和 pSic1 合奏的微扰影响很小。此外,与如此多样化的实验数据集的相互一致表明,现在可以高度自信地检查生成的集合的细节,例如整体紧凑性和端到端距离波动,以揭示 Sic1 的显着特征与 pSic1 相比。从实验得到很好支持的合奏中,我们发现它们与 Sic1' 的独立生物物理模型一致 s 与其合作伙伴 Cdc4 的超灵敏结合。我们的结果强调了综合建模和验证在从 IDP 构象集合计算和得出生物学结论中的重要性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号