European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2020-08-21 , DOI: 10.1016/j.ejmech.2020.112752 Huda K Mahmoud 1 , Thoraya A Farghaly 2 , Hanan G Abdulwahab 3 , Nadia T Al-Qurashi 4 , Mohamed R Shaaban 2
|
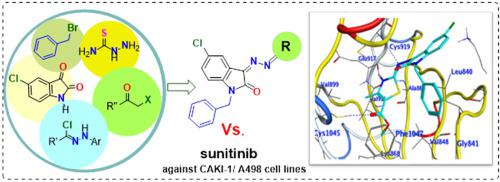
Novel 2-indolinone thiazole hybrids were designed and synthesized as VEGFR-2 inhibitors based on sunitinib, an FDA-approved anticancer drug. The proposed structures of the prepared 2-indolinone thiazole hybrids were confirmed based on their spectral data and CHN analyses. The target compounds were screened in vitro for their anti-VEGFR-2 activity. All tested compounds exhibited a potent submicromolar inhibition of VEGFR-2 kinase with IC50 values ranging from 0.067 to 0.422 μM, relative to sunitinib reference drug (IC50 = 0.075 ± 0.002 μM). Compounds 5, 15a, 15b, 17, 19c displayed excellent VEGFR-2 inhibitory activity, comparable or nearly equipotent to sunitinib. Compound 13b stood out as the most potent against VEGFR-2 showing IC50 value of 0.067 ± 0.002 μM, lower than that of sunitinib. In addition, the most potent derivatives were assessed for their anticancer activity against two renal cancer cell lines. Compound 13b (IC50 = 3.9 ± 0.13 μM) was more potent than sunitinib (IC50 = 4.93 ± 0.16 μM) against CAKI-1 cell line. Moreover, thiazole 15b displayed excellent anticancer activity against CAKI-1 cell line (IC50 = 3.31 ± 0.11 μM), superior to that of sunitinib (IC50 = 4.93 ± 0.16 μM). Thiazole 15b was also equipotent to sunitinib (IC50 = 1.23 ± 0.04 μM) against A498 cell line. Besides, compound 15b revealed a safety profile much better than that of sunitinib against normal human renal cells. Furthermore, a docking study revealed a proper fitting of the most active compounds into the ATP binding site of VEGFR-2, rationalizing their potent anti-VEGFR-2 activity.
中文翻译:
新型2-吲哚啉酮噻唑杂化物作为舒尼替尼类似物:设计,合成和有效的VEGFR-2抑制作用,具有潜在的抗肾癌活性。
根据FDA批准的抗癌药舒尼替尼,设计并合成了新型2-吲哚酮噻唑杂合物作为VEGFR-2抑制剂。基于其光谱数据和CHN分析,确定了所制备的2-吲哚酮噻唑杂化物的拟议结构。体外筛选目标化合物的抗VEGFR-2活性。相对于舒尼替尼参考药物(IC 50 = 0.075±0.002μM),所有测试的化合物均表现出对VEGFR-2激酶的有效的亚微摩尔抑制作用,IC 50值为0.067至0.422μM 。化合物5、15a,15b,17、19c显示出优异的VEGFR-2抑制活性,与舒尼替尼相当或几乎等效。化合物13b在对抗VEGFR-2方面表现最强,IC 50值为0.067±0.002μM,低于舒尼替尼。另外,评估了最有效的衍生物对两种肾癌细胞系的抗癌活性。 对于CAKI-1细胞系,化合物13b(IC 50 = 3.9± 0.13μM)比舒尼替尼(IC 50 = 4.93±0.16μM )更有效。此外,噻唑15b对CAKI-1细胞系具有出色的抗癌活性(IC 50 = 3.31± 0.11μM ),优于舒尼替尼(IC 50 = 4.93±0.16μM)。噻唑15b也等同于舒尼替尼(IC 50 = 1.23±0.04μM)。此外,化合物15b显示出比舒尼替尼对正常人肾细胞的安全性要好得多。此外,对接研究显示,活性最高的化合物可以适当地适合VEGFR-2的ATP结合位点,从而使其有效的抗VEGFR-2活性合理化。










































 京公网安备 11010802027423号
京公网安备 11010802027423号