当前位置:
X-MOL 学术
›
Dalton Trans.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
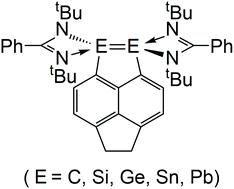
A theoretical study of the reactivity of ethene and benzophenone with a hyper-coordinated alkene containing a so-called E=E (E = C, Si, Ge, Sn, and Pb) unit.
Dalton Transactions ( IF 3.5 ) Pub Date : 2020-08-19 , DOI: 10.1039/d0dt01914c Ming-Chung Yang,Ming-Der Su
Dalton Transactions ( IF 3.5 ) Pub Date : 2020-08-19 , DOI: 10.1039/d0dt01914c Ming-Chung Yang,Ming-Der Su
|
The reactivity of a reported hyper-coordinated alkene (Rea-E; Rea = reactant; E = group 14 element) featuring a central E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E moiety was theoretically analyzed using DFT (density functional theory) and the EDA-NOCV (energy decomposition analysis–natural orbitals for chemical valence) method. M06-2X/def2-SVP and B3LYP-D3/def2-SVP results demonstrate that five Rea-E molecules have an energy minimum as their structures have no imaginary frequency. Theoretical examinations based on three types of bond order calculations (Wiberg, Mayer, and Fuzzy), the LOL (localized orbital locator) analyses, Lewis structures and the NBO (natural bond orbital) analyses suggest that a very weak central Si–Si single bond and an extremely weak central Ge–Ge single bond, rather than a double bond, are present in the Rea-Si and Rea-Ge molecules, respectively. On the other hand, no bond is found between the two central group 14 atoms in Rea-C, Rea-Sn, and Rea-Pb. The theoretical investigation demonstrates that the reactivity of the Rea-E compound decreases in the order Rea-Si > Rea-Ge > Rea-C, a trend that results from the differences in the atomic radii of the group 14 elements. Carbon has the smallest atomic radius in the group 14 family, causing steric crowding between Rea-C and other attacking species. This circumstance, in turn, increases the activation energies of its addition reactions and renders these reactions energetically infeasible. For the cyclic product of Rea-Ge, the theoretical evidence reveals that the comparatively large atomic radius of Ge induces the weakest Pauli repulsions and the smallest overlap integrals between Rea-Ge and the other doubly bonded molecules. This situation, in turn, makes the overall cyclization reaction of Rea-Ge endothermic. As a result, only the silicon-centered molecule, Rea-Si, can undergo the [2 + 2] cycloaddition reactions with doubly bonded molecules without kinetic or thermodynamic difficulty, which agrees well with the available experimental findings.
E moiety was theoretically analyzed using DFT (density functional theory) and the EDA-NOCV (energy decomposition analysis–natural orbitals for chemical valence) method. M06-2X/def2-SVP and B3LYP-D3/def2-SVP results demonstrate that five Rea-E molecules have an energy minimum as their structures have no imaginary frequency. Theoretical examinations based on three types of bond order calculations (Wiberg, Mayer, and Fuzzy), the LOL (localized orbital locator) analyses, Lewis structures and the NBO (natural bond orbital) analyses suggest that a very weak central Si–Si single bond and an extremely weak central Ge–Ge single bond, rather than a double bond, are present in the Rea-Si and Rea-Ge molecules, respectively. On the other hand, no bond is found between the two central group 14 atoms in Rea-C, Rea-Sn, and Rea-Pb. The theoretical investigation demonstrates that the reactivity of the Rea-E compound decreases in the order Rea-Si > Rea-Ge > Rea-C, a trend that results from the differences in the atomic radii of the group 14 elements. Carbon has the smallest atomic radius in the group 14 family, causing steric crowding between Rea-C and other attacking species. This circumstance, in turn, increases the activation energies of its addition reactions and renders these reactions energetically infeasible. For the cyclic product of Rea-Ge, the theoretical evidence reveals that the comparatively large atomic radius of Ge induces the weakest Pauli repulsions and the smallest overlap integrals between Rea-Ge and the other doubly bonded molecules. This situation, in turn, makes the overall cyclization reaction of Rea-Ge endothermic. As a result, only the silicon-centered molecule, Rea-Si, can undergo the [2 + 2] cycloaddition reactions with doubly bonded molecules without kinetic or thermodynamic difficulty, which agrees well with the available experimental findings.
中文翻译:

乙烯和二苯甲酮与含有所谓E = E(E = C,Si,Ge,Sn和Pb)单元的超配位烯烃的反应性的理论研究。
使用DFT(密度泛函理论)和EDA-NOCV(能量分解分析,理论上分析了已报道的具有中心E E部分的超配位烯烃(Rea-E; Rea =反应物; E =组14元素)的反应性化学价的自然轨道)方法。M06-2X / def2-SVP和B3LYP-D3 / def2-SVP结果证明五个Rea-E由于分子的结构没有虚数频率,因此它们的能量最小。基于三种键序计算(Wiberg,Mayer和Fuzzy),LOL(局部轨道定位器)分析,Lewis结构和NBO(自然键轨道)分析的理论检验表明,中心Si-Si单键非常弱Rea-Si和Rea-Ge分子中分别存在一个非常弱的中心Ge-Ge单键而不是双键。另一方面,在Rea-C,Rea-Sn和Rea-Pb中的两个中心基团14原子之间未发现键。理论研究表明,Rea-E的反应活性化合物以Rea-Si > Rea-Ge > Rea-C的顺序减少,这种趋势是由第14组元素的原子半径不同引起的。碳在第14组族中的原子半径最小,导致Rea-C与其他攻击物种之间的空间拥挤。这种情况继而增加了其加成反应的活化能,并使这些反应在能量上不可行。对于Rea-Ge的循环产物,理论证据表明,Ge的相对较大的原子半径会引起Rea-Ge之间最弱的Pauli排斥力和最小的重叠积分。和其他双键分子。反过来,这种情况使Rea-Ge的整体环化反应发生吸热。结果,只有以硅为中心的分子Rea-Si可以与双键分子进行[2 + 2]环加成反应,而没有动力学或热力学困难,这与现有的实验结果十分吻合。
更新日期:2020-09-22
E moiety was theoretically analyzed using DFT (density functional theory) and the EDA-NOCV (energy decomposition analysis–natural orbitals for chemical valence) method. M06-2X/def2-SVP and B3LYP-D3/def2-SVP results demonstrate that five Rea-E molecules have an energy minimum as their structures have no imaginary frequency. Theoretical examinations based on three types of bond order calculations (Wiberg, Mayer, and Fuzzy), the LOL (localized orbital locator) analyses, Lewis structures and the NBO (natural bond orbital) analyses suggest that a very weak central Si–Si single bond and an extremely weak central Ge–Ge single bond, rather than a double bond, are present in the Rea-Si and Rea-Ge molecules, respectively. On the other hand, no bond is found between the two central group 14 atoms in Rea-C, Rea-Sn, and Rea-Pb. The theoretical investigation demonstrates that the reactivity of the Rea-E compound decreases in the order Rea-Si > Rea-Ge > Rea-C, a trend that results from the differences in the atomic radii of the group 14 elements. Carbon has the smallest atomic radius in the group 14 family, causing steric crowding between Rea-C and other attacking species. This circumstance, in turn, increases the activation energies of its addition reactions and renders these reactions energetically infeasible. For the cyclic product of Rea-Ge, the theoretical evidence reveals that the comparatively large atomic radius of Ge induces the weakest Pauli repulsions and the smallest overlap integrals between Rea-Ge and the other doubly bonded molecules. This situation, in turn, makes the overall cyclization reaction of Rea-Ge endothermic. As a result, only the silicon-centered molecule, Rea-Si, can undergo the [2 + 2] cycloaddition reactions with doubly bonded molecules without kinetic or thermodynamic difficulty, which agrees well with the available experimental findings.
中文翻译:
乙烯和二苯甲酮与含有所谓E = E(E = C,Si,Ge,Sn和Pb)单元的超配位烯烃的反应性的理论研究。
使用DFT(密度泛函理论)和EDA-NOCV(能量分解分析,理论上分析了已报道的具有中心E E部分的超配位烯烃(Rea-E; Rea =反应物; E =组14元素)的反应性
化学价的自然轨道)方法。M06-2X / def2-SVP和B3LYP-D3 / def2-SVP结果证明五个Rea-E由于分子的结构没有虚数频率,因此它们的能量最小。基于三种键序计算(Wiberg,Mayer和Fuzzy),LOL(局部轨道定位器)分析,Lewis结构和NBO(自然键轨道)分析的理论检验表明,中心Si-Si单键非常弱Rea-Si和Rea-Ge分子中分别存在一个非常弱的中心Ge-Ge单键而不是双键。另一方面,在Rea-C,Rea-Sn和Rea-Pb中的两个中心基团14原子之间未发现键。理论研究表明,Rea-E的反应活性化合物以Rea-Si > Rea-Ge > Rea-C的顺序减少,这种趋势是由第14组元素的原子半径不同引起的。碳在第14组族中的原子半径最小,导致Rea-C与其他攻击物种之间的空间拥挤。这种情况继而增加了其加成反应的活化能,并使这些反应在能量上不可行。对于Rea-Ge的循环产物,理论证据表明,Ge的相对较大的原子半径会引起Rea-Ge之间最弱的Pauli排斥力和最小的重叠积分。和其他双键分子。反过来,这种情况使Rea-Ge的整体环化反应发生吸热。结果,只有以硅为中心的分子Rea-Si可以与双键分子进行[2 + 2]环加成反应,而没有动力学或热力学困难,这与现有的实验结果十分吻合。








































 京公网安备 11010802027423号
京公网安备 11010802027423号