Journal of Molecular Graphics and Modelling ( IF 2.7 ) Pub Date : 2020-08-15 , DOI: 10.1016/j.jmgm.2020.107713 George Baffour Pipim 1 , Richard Tia 1 , Evans Adei 1
|
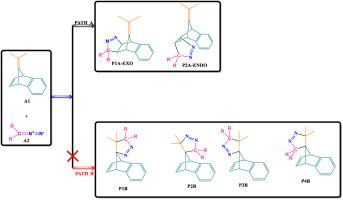
The ability to synthesize targeted molecules hinges on detailed mechanistic insight of the reaction. The 1,3-dipolar cycloaddition reaction between diazomethane derivatives and 7-isopropylidenebenzonorbornadiene have been extensively studied using density functional theory (DFT) at the M06–2X/6-311G(d,p) level of theory in order to delineate the peri-, regio-, and stereo-selectivities of the reaction. The diazomethane is shown to periselectively add across the endocyclic olefinic bond of the 7-isopropylidenebenzonorbornadiene and stereoselectively in the exo fashion, yielding the exo-cycloadduct as the major product, with a rate constant of 3.83 × 104 s−1. The endo approach of this periselective path is the closest competing pathway with a rate constant of 8.78 × 101 s−1. Neither electron-donating groups (R = methyl, ethyl, amine, cyclopropyl) nor electron-withdrawing groups (R = cyano, nitro, carbonyl) on the diazomethane alters the peri- and stereo-selectivity of the reaction. However, the substituents do have an effect on whether the addition follow normal or inverse electron demand mechanisms. EDGs favor a normal electron demand mechanism while EWGs favor an inverse electron demand 1,3-dipolar cycloaddition reaction. While EDGs-substituted diazomethane derivatives behave as nucleophiles in reactions with 7-isopropylidenebenzonorbornadiene, EWGs-substituted diazomethane derivatives behave as electrophiles. The 1,3-dipole adds across the dipolarophile via a concerted asynchronous mechanism, but a stepwise diradical mechanism has been ruled out. The selectivities observed in the title reaction are kinetically controlled. Analysis of the nucleophilic Parr function ( at the different reaction sites in the dipolarophile indicates that the diazomethane adds across the atomic centers with highest NBO and Mulliken atomic spin densities.
中文翻译:
7-异亚丙基苯并降冰片二烯与重氮甲烷衍生物的(3 + 2)环加成反应:理论研究。
合成目标分子的能力取决于反应的详细机理。重氮甲烷衍生物与7-异亚丙基苯并降冰片二烯之间的1,3-偶极环加成反应已在M06–2X / 6-311G(d,p)的理论水平上使用密度泛函理论(DFT)进行了广泛研究,以描绘出反应的区域,区域和立体选择性。重氮甲烷显示出选择性地跨7-异亚丙基苯并降冰片二烯的内环烯烃键加成,并以exo方式立体选择性地生成,以exo- cycloadduct为主要产物,速率常数为3.83×10 4 s -1。这种围选择性途径的内在途径是最接近的竞争途径,其速率常数为8.78×10 1 s -1。重氮甲烷上的给电子基团(R =甲基,乙基,胺,环丙基)或吸电子基团(R =氰基,硝基,羰基)都不会改变反应的选择性和立体选择性。但是,取代基确实对加成是否遵循正电子或逆电子需求机制有影响。EDG支持正常的电子需求机制,而EWG支持逆电子需求的1,3-偶极环加成反应。尽管EDGs取代的重氮甲烷衍生物在与7-异亚丙基苯并降冰片二烯的反应中起亲核试剂的作用,但EWGs取代的重氮甲烷衍生物在亲电试剂中的作用。1,3-偶极子通过协同异步机制跨亲核子增加,但已排除了逐步的双自由基机制。标题反应中观察到的选择性是动力学控制的。亲核Parr功能分析( 在双极亲子分子的不同反应位点处,表明重氮甲烷以最高的NBO和Mulliken原子自旋密度跨原子中心添加。










































 京公网安备 11010802027423号
京公网安备 11010802027423号