当前位置:
X-MOL 学术
›
Angew. Chem. Int. Ed.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Co-crystal Prediction by Artificial Neural Networks.
Angewandte Chemie International Edition ( IF 16.1 ) Pub Date : 2020-08-14 , DOI: 10.1002/anie.202009467 Jan-Joris Devogelaer 1 , Hugo Meekes 1 , Paul Tinnemans 1 , Elias Vlieg 1 , René de Gelder 1
Angewandte Chemie International Edition ( IF 16.1 ) Pub Date : 2020-08-14 , DOI: 10.1002/anie.202009467 Jan-Joris Devogelaer 1 , Hugo Meekes 1 , Paul Tinnemans 1 , Elias Vlieg 1 , René de Gelder 1
Affiliation
|
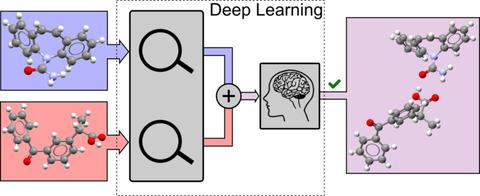
A significant amount of attention has been given to the design and synthesis of co‐crystals by both industry and academia because of its potential to change a molecule's physicochemical properties. Yet, difficulties arise when searching for adequate combinations of molecules (or coformers) to form co‐crystals, hampering the efficient exploration of the target's solid‐state landscape. This paper reports on the application of a data‐driven co‐crystal prediction method based on two types of artificial neural network models and co‐crystal data present in the Cambridge Structural Database. The models accept pairs of coformers and predict whether a co‐crystal is likely to form. By combining the output of multiple models of both types, our approach shows to have excellent performance on the proposed co‐crystal training and validation sets, and has an estimated accuracy of 80 % for molecules for which previous co‐crystallization data is unavailable.
中文翻译:

人工神经网络的共晶预测。
由于共晶具有改变分子物理化学性质的潜力,工业界和学术界都对共晶的设计和合成给予了极大的关注。然而,在寻找足够的分子(或共形成体)组合以形成共晶时会出现困难,从而阻碍了对目标固态景观的有效探索。本文报告了基于两种类型的人工神经网络模型和剑桥结构数据库中存在的共晶数据的数据驱动的共晶预测方法的应用。该模型接受成对的共晶并预测是否可能形成共晶。通过结合两种类型的多个模型的输出,我们的方法在所提出的共晶训练和验证集上表现出优异的性能,并且对于先前无法获得共晶数据的分子,估计准确度为 80%。
更新日期:2020-08-14
中文翻译:
人工神经网络的共晶预测。
由于共晶具有改变分子物理化学性质的潜力,工业界和学术界都对共晶的设计和合成给予了极大的关注。然而,在寻找足够的分子(或共形成体)组合以形成共晶时会出现困难,从而阻碍了对目标固态景观的有效探索。本文报告了基于两种类型的人工神经网络模型和剑桥结构数据库中存在的共晶数据的数据驱动的共晶预测方法的应用。该模型接受成对的共晶并预测是否可能形成共晶。通过结合两种类型的多个模型的输出,我们的方法在所提出的共晶训练和验证集上表现出优异的性能,并且对于先前无法获得共晶数据的分子,估计准确度为 80%。










































 京公网安备 11010802027423号
京公网安备 11010802027423号