Current Topics in Medicinal Chemistry ( IF 2.9 ) Pub Date : 2020-08-31 , DOI: 10.2174/1568026620666200704135327 Anuraj Nayarisseri 1, 2, 3, 4 , Ravina Khandelwal 1 , Maddala Madhavi 5 , Chandrabose Selvaraj 4 , Umesh Panwar 4 , Khushboo Sharma 1 , Tajamul Hussain 3, 6 , Sanjeev Kumar Singh 4
|
Background: The vast geographical expansion of novel coronavirus and an increasing number of COVID-19 affected cases have overwhelmed health and public health services. Artificial Intelligence (AI) and Machine Learning (ML) algorithms have extended their major role in tracking disease patterns, and in identifying possible treatments.
Objective: This study aims to identify potential COVID-19 protease inhibitors through shape-based Machine Learning assisted by Molecular Docking and Molecular Dynamics simulations.
Methods: 31 Repurposed compounds have been selected targeting the main coronavirus protease (6LU7) and a machine learning approach was employed to generate shape-based molecules starting from the 3D shape to the pharmacophoric features of their seed compound. Ligand-Receptor Docking was performed with Optimized Potential for Liquid Simulations (OPLS) algorithms to identify highaffinity compounds from the list of selected candidates for 6LU7, which were subjected to Molecular Dynamic Simulations followed by ADMET studies and other analyses.
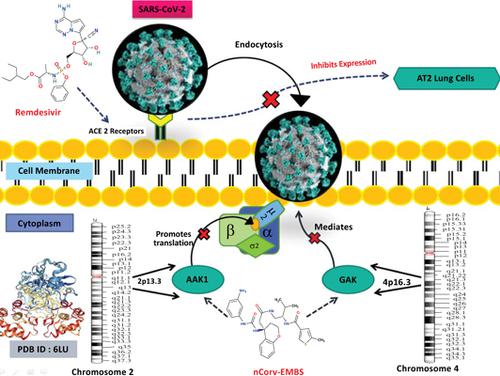
Results: Shape-based Machine learning reported remdesivir, valrubicin, aprepitant, and fulvestrant as the best therapeutic agents with the highest affinity for the target protein. Among the best shape-based compounds, a novel compound identified was not indexed in any chemical databases (PubChem, Zinc, or ChEMBL). Hence, the novel compound was named 'nCorv-EMBS'. Further, toxicity analysis showed nCorv-EMBS to be suitable for further consideration as the main protease inhibitor in COVID-19.
Conclusion: Effective ACE-II, GAK, AAK1, and protease 3C blockers can serve as a novel therapeutic approach to block the binding and attachment of the main COVID-19 protease (PDB ID: 6LU7) to the host cell and thus inhibit the infection at AT2 receptors in the lung. The novel compound nCorv- EMBS herein proposed stands as a promising inhibitor to be evaluated further for COVID-19 treatment.
中文翻译:
分子动力学模拟辅助的潜在新型COVID-19蛋白酶抑制剂的基于形状的机器学习模型。
背景:新型冠状病毒的广泛地域扩张和受COVID-19感染的病例数量增加,已经使卫生和公共卫生服务不堪重负。人工智能(AI)和机器学习(ML)算法扩展了其在跟踪疾病模式和确定可能的治疗方法中的主要作用。
目的:本研究旨在通过基于分子对接和分子动力学模拟的基于形状的机器学习来识别潜在的COVID-19蛋白酶抑制剂。
方法:已经选择了31种针对主要冠状病毒蛋白酶(6LU7)的目标化合物,并采用了机器学习方法来生成基于形状的分子,从3D形状到其种子化合物的药效学特征。配体-受体对接是通过优化的液体模拟电位(OPLS)算法进行的,以从6LU7的选定候选物列表中识别高亲和力化合物,然后对这些化合物进行分子动力学模拟,然后进行ADMET研究和其他分析。
结果:基于形状的机器学习报告瑞姆昔韦,缬卢比,阿瑞匹坦和氟维司群是对靶蛋白具有最高亲和力的最佳治疗剂。在最佳的基于形状的化合物中,已鉴定的新化合物未在任何化学数据库(PubChem,Zinc或ChEMBL)中建立索引。因此,该新型化合物被命名为“ nCorv-EMBS”。此外,毒性分析表明,nCorv-EMBS适合作为COVID-19中的主要蛋白酶抑制剂进行进一步研究。
结论:有效的ACE-II,GAK,AAK1和蛋白酶3C阻断剂可作为一种新的治疗方法,阻断主要COVID-19蛋白酶(PDB ID:6LU7)与宿主细胞的结合和附着,从而抑制感染在肺中的AT2受体上。本文提出的新型化合物nCorv-EMBS是有前途的抑制剂,需要进一步评估以用于COVID-19治疗。










































 京公网安备 11010802027423号
京公网安备 11010802027423号