当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
MARTINI-Compatible Coarse-Grained Model for the Mesoscale Simulation of Peptoids.
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2020-08-10 , DOI: 10.1021/acs.jpcb.0c04567 Mingfei Zhao 1 , Janani Sampath 2 , Sarah Alamdari 3 , Gillian Shen 4 , Chun-Long Chen 2, 3 , Christopher J Mundy 2, 3 , Jim Pfaendtner 2, 3 , Andrew L Ferguson 1
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2020-08-10 , DOI: 10.1021/acs.jpcb.0c04567 Mingfei Zhao 1 , Janani Sampath 2 , Sarah Alamdari 3 , Gillian Shen 4 , Chun-Long Chen 2, 3 , Christopher J Mundy 2, 3 , Jim Pfaendtner 2, 3 , Andrew L Ferguson 1
Affiliation
|
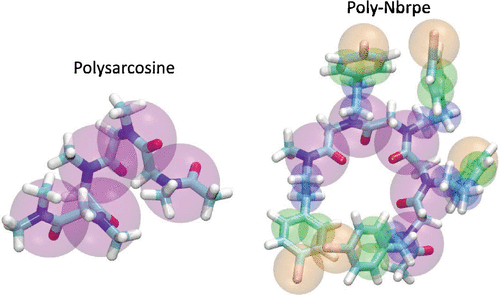
Peptoids (poly-N-substituted glycines) are a class of synthetic polymers that are regioisomers of peptides (poly-C-substituted glycines), in which the point of side-chain connectivity is shifted from the backbone C to the N atom. Peptoids have found diverse applications as peptidomimetic drugs, protein mimetic polymers, surfactants, and catalysts. Computational modeling is valuable in the understanding and design of peptoid-based nanomaterials. In this work, we report the bottom-up parameterization of coarse-grained peptoid force fields based on the MARTINI peptide force field against all-atom peptoid simulation data. Our parameterization pipeline iteratively refits coarse-grained bonded interactions using iterative Boltzmann inversion and nonbonded interactions by matching the potential of mean force for chain extension. We assure good sampling of the amide bond cis/trans isomerizations in the all-atom simulation data using parallel bias metadynamics. We develop coarse-grained models for two representative peptoids—polysarcosine (poly(N-methyl glycine)) and poly(N-((4-bromophenyl)ethyl)glycine)—and show their structural and thermodynamic properties to be in excellent accord with all-atom calculations but up to 25-fold more efficient and compatible with MARTINI force fields. This work establishes a new rigorously parameterized coarse-grained peptoid force field for the understanding and design of peptoid nanomaterials at length and time scales inaccessible to all-atom calculations.
中文翻译:

类肽的中尺度模拟的兼容MARTINI的粗粒度模型。
类肽(聚-N-取代的甘氨酸)是一类合成的聚合物,它们是肽的区域异构体(聚-C-取代的甘氨酸),其中侧链连接点从主链C移至N原子。拟肽已被用作拟肽药物,模拟蛋白质的聚合物,表面活性剂和催化剂的多种应用。计算模型对于基于类肽的纳米材料的理解和设计非常有价值。在这项工作中,我们报告了针对所有原子类肽模拟数据的基于MARTINI肽力场的自下而上的粗粒类肽力场的参数化。我们的参数化管道使用迭代玻尔兹曼反演和非键合相互作用迭代匹配粗粒键合相互作用,方法是匹配平均力链扩展的潜力。我们确保使用平行偏置元动力学在所有原子的模拟数据中对酰胺键的顺/反异构化进行良好采样。我们开发了两种代表性类肽-粗肌氨酸(poly(N-甲基甘氨酸))和聚(N -((4-溴苯基)乙基)甘氨酸)的结构和热力学性质与全原子计算非常吻合,但效率高达25倍,并且与MARTINI力场。这项工作建立了一个新的严格参数化的粗粒类肽作用力场,用于了解和设计类肽纳米材料的长度和时间尺度,而这是所有原子计算无法获得的。
更新日期:2020-09-10
中文翻译:
类肽的中尺度模拟的兼容MARTINI的粗粒度模型。
类肽(聚-N-取代的甘氨酸)是一类合成的聚合物,它们是肽的区域异构体(聚-C-取代的甘氨酸),其中侧链连接点从主链C移至N原子。拟肽已被用作拟肽药物,模拟蛋白质的聚合物,表面活性剂和催化剂的多种应用。计算模型对于基于类肽的纳米材料的理解和设计非常有价值。在这项工作中,我们报告了针对所有原子类肽模拟数据的基于MARTINI肽力场的自下而上的粗粒类肽力场的参数化。我们的参数化管道使用迭代玻尔兹曼反演和非键合相互作用迭代匹配粗粒键合相互作用,方法是匹配平均力链扩展的潜力。我们确保使用平行偏置元动力学在所有原子的模拟数据中对酰胺键的顺/反异构化进行良好采样。我们开发了两种代表性类肽-粗肌氨酸(poly(N-甲基甘氨酸))和聚(N -((4-溴苯基)乙基)甘氨酸)的结构和热力学性质与全原子计算非常吻合,但效率高达25倍,并且与MARTINI力场。这项工作建立了一个新的严格参数化的粗粒类肽作用力场,用于了解和设计类肽纳米材料的长度和时间尺度,而这是所有原子计算无法获得的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号