Journal of Molecular Biology ( IF 5.6 ) Pub Date : 2020-08-06 , DOI: 10.1016/j.jmb.2020.07.027 Biao Zhang 1 , Xi Zhang 2 , Robin Pearce 2 , Hong-Bin Shen 3 , Yang Zhang 4
|
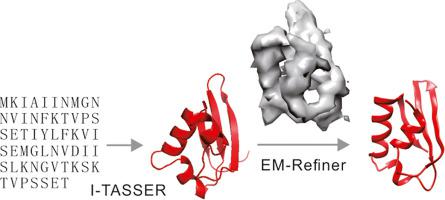
The rapid progress of cryo-electron microscopy (cryo-EM) in structural biology has raised an urgent need for robust methods to create and refine atomic-level structural models using low-resolution EM density maps. We propose a new protocol to create initial models using I-TASSER protein structure prediction, followed by EM density map-based rigid-body structure fitting, flexible fragment adjustment and atomic-level structure refinement simulations. The protocol was tested on a large set of 285 non-homologous proteins and generated structural models with correct folds for 260 proteins, where 28% had RMSDs below 2 Å. Compared to other state-of-the-art methods, the major advantage of the proposed pipeline lies in the uniform structure prediction and refinement protocol, as well as the extensive structural re-assembly simulations, which allow for low-to-medium resolution EM density map-guided structure modeling starting from amino acid sequences. Interestingly, the quality of both the image fitting and subsequent structure refinement was found to be strongly correlated with the correctness of the initial I-TASSER models; this is mainly due to the different correlation patterns observed between force field and structural quality for the models with template modeling score (or TM-score, a metric quantifying the similarity of models to the native) above and below a threshold of 0.5. Overall, the results demonstrate a new avenue that is ready to use for large-scale cryo-EM-based structure modeling and atomic-level density map-guided structure refinement.
中文翻译:
使用中低分辨率冷冻电镜密度图进行原子级蛋白质结构建模和细化的新方案。
结构生物学中低温电子显微镜 (cryo-EM) 的快速发展迫切需要使用低分辨率 EM 密度图创建和完善原子级结构模型的稳健方法。我们提出了一种使用 I-TASSER 蛋白质结构预测创建初始模型的新协议,然后是基于 EM 密度图的刚体结构拟合、灵活的片段调整和原子级结构细化模拟。该协议在大量 285 种非同源蛋白质上进行了测试,并生成了具有 260 种蛋白质正确折叠的结构模型,其中 28% 的 RMSD 低于 2 Å。与其他最先进的方法相比,所提出的管道的主要优势在于统一的结构预测和细化协议,以及广泛的结构重新组装模拟,允许从氨基酸序列开始进行中低分辨率 EM 密度图引导的结构建模。有趣的是,发现图像拟合和后续结构细化的质量与初始 I-TASSER 模型的正确性密切相关;这主要是由于模板建模分数(或 TM 分数,一种量化模型与本地模型相似性的度量)高于和低于 0.5 阈值的模型在力场和结构质量之间观察到不同的相关模式。总体而言,结果展示了一条新的途径,可用于大规模的基于低温电磁学的结构建模和原子级密度图引导的结构细化。发现图像拟合和随后的结构细化的质量与初始 I-TASSER 模型的正确性密切相关;这主要是由于模板建模分数(或 TM 分数,一种量化模型与原生模型相似性的度量)高于和低于 0.5 阈值的模型在力场和结构质量之间观察到不同的相关模式。总体而言,结果展示了一条新的途径,可用于大规模的基于低温电磁学的结构建模和原子级密度图引导的结构细化。发现图像拟合和随后的结构细化的质量与初始 I-TASSER 模型的正确性密切相关;这主要是由于模板建模分数(或 TM 分数,一种量化模型与原生模型相似性的度量)高于和低于 0.5 阈值的模型在力场和结构质量之间观察到不同的相关模式。总体而言,结果展示了一条新的途径,可用于大规模的基于低温电磁学的结构建模和原子级密度图引导的结构细化。这主要是由于模板建模分数(或 TM 分数,一种量化模型与原生模型相似性的度量)高于和低于 0.5 阈值的模型在力场和结构质量之间观察到不同的相关模式。总体而言,结果展示了一种新的途径,可用于大规模的基于低温 EM 的结构建模和原子级密度图引导的结构细化。这主要是由于模板建模分数(或 TM 分数,一种量化模型与原生模型相似性的度量)高于和低于 0.5 阈值的模型在力场和结构质量之间观察到不同的相关模式。总体而言,结果展示了一条新的途径,可用于大规模的基于低温电磁学的结构建模和原子级密度图引导的结构细化。


























 京公网安备 11010802027423号
京公网安备 11010802027423号