当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Kinetic modeling of methyl pentanoate pyrolysis based on ab initio calculations.
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2020-07-30 , DOI: 10.1039/d0cp02821e Yanlei Shang 1 , Hongbo Ning 1 , Jinchun Shi 2 , Sheng-Nian Luo 1
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2020-07-30 , DOI: 10.1039/d0cp02821e Yanlei Shang 1 , Hongbo Ning 1 , Jinchun Shi 2 , Sheng-Nian Luo 1
Affiliation
|
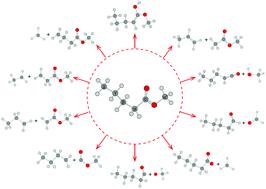
Recently, methyl pentanoate (MP) was proposed as a viable biodiesel surrogate to petroleum-based fuels. To better understand the pyrolysis chemistry of MP, the unimolecular decomposition kinetics of MP is theoretically investigated on the basis of ab initio calculations; ten primary channels, including four intramolecular H-shifts and six C–C and C–O bond fissions, are identified. The geometries are optimized at the M06-2X/cc-pVTZ level of theory, and accurate barrier heights are determined using the DLPNO-CCSD(T)/CBS(T-Q) method, which shows a good performance against the CCSD(T)/CBS(T-Q) method with an uncertainty of 0.5 kcal mol−1 for small methyl esters. The atomization enthalpy method is adopted to obtain the thermodynamics of involved species. The Rice–Ramsperger–Kassel–Marcus/master equation theory coupled with one-dimensional hindered rotor approximation is employed to calculate the phenomenological rate constants at 500–2000 K and 0.01–100 atm. The branching ratio analysis indicates that two reactions, MP ↔ CH3OC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CH3 + CH2CHCH3 and MP ↔ CH3OC(O)CH2 + CH2CH2CH3, are the dominant channels at low and high temperatures, respectively. The model from Diévart et al. [Proc. Combust. Inst., 2013, 34(1), 821–829] is updated with our calculations, and the modified model can yield a better prediction in reproducing the ignition delay times of MP at high temperatures. This work provides a comprehensive investigation of MP unimolecular decomposition, and can serve as a prototype for understanding the pyrolysis of larger alkyl esters.
O)CH3 + CH2CHCH3 and MP ↔ CH3OC(O)CH2 + CH2CH2CH3, are the dominant channels at low and high temperatures, respectively. The model from Diévart et al. [Proc. Combust. Inst., 2013, 34(1), 821–829] is updated with our calculations, and the modified model can yield a better prediction in reproducing the ignition delay times of MP at high temperatures. This work provides a comprehensive investigation of MP unimolecular decomposition, and can serve as a prototype for understanding the pyrolysis of larger alkyl esters.
中文翻译:

基于从头算的戊酸甲酯热解动力学模型。
最近,已提出戊酸甲酯(MP)作为石油基燃料的可行生物柴油替代品。为了更好地理解MP的热解化学,理论上根据从头算的方法研究了MP的单分子分解动力学。确定了十个主要通道,包括四个分子内H位移和六个C–C和C–O键裂变。在M06-2X / cc-pVTZ的理论水平上优化了几何形状,并使用DLPNO-CCSD(T)/ CBS(TQ)方法确定了准确的势垒高度,相对于CCSD(T)/不确定度为0.5 kcal mol -1的CBS(TQ)方法用于小的甲酯。采用雾化焓法获得所涉及物质的热力学。Rice-Ramsperger-Kassel-Marcus /主方程理论与一维受阻转子逼近一起用于计算500-2000 K和0.01-100 atm的现象速率常数。支化比分析表明,MP↔CH 3 OC(O)CH 3 + CH 2 CHCH 3和MP↔CH 3 OC(O)CH 2 + CH 2 CH 2 CH 3是两个反应的主要反应通道。高温。Diévart等人的模型。[进程 燃烧 研究所 ,2013,34(1),821-829]与我们的计算更新,并且修改后的模型可以在高温下再现MP的点火延迟时间产生更好的预测。这项工作提供了MP单分子分解的全面研究,并可以作为了解更大的烷基酯热解的原型。
更新日期:2020-08-25
O)CH3 + CH2CHCH3 and MP ↔ CH3OC(O)CH2 + CH2CH2CH3, are the dominant channels at low and high temperatures, respectively. The model from Diévart et al. [Proc. Combust. Inst., 2013, 34(1), 821–829] is updated with our calculations, and the modified model can yield a better prediction in reproducing the ignition delay times of MP at high temperatures. This work provides a comprehensive investigation of MP unimolecular decomposition, and can serve as a prototype for understanding the pyrolysis of larger alkyl esters.
中文翻译:
基于从头算的戊酸甲酯热解动力学模型。
最近,已提出戊酸甲酯(MP)作为石油基燃料的可行生物柴油替代品。为了更好地理解MP的热解化学,理论上根据从头算的方法研究了MP的单分子分解动力学。确定了十个主要通道,包括四个分子内H位移和六个C–C和C–O键裂变。在M06-2X / cc-pVTZ的理论水平上优化了几何形状,并使用DLPNO-CCSD(T)/ CBS(TQ)方法确定了准确的势垒高度,相对于CCSD(T)/不确定度为0.5 kcal mol -1的CBS(TQ)方法用于小的甲酯。采用雾化焓法获得所涉及物质的热力学。Rice-Ramsperger-Kassel-Marcus /主方程理论与一维受阻转子逼近一起用于计算500-2000 K和0.01-100 atm的现象速率常数。支化比分析表明,MP↔CH 3 OC(
O)CH 3 + CH 2 CHCH 3和MP↔CH 3 OC(O)CH 2 + CH 2 CH 2 CH 3是两个反应的主要反应通道。高温。Diévart等人的模型。[进程 燃烧 研究所 ,2013,34(1),821-829]与我们的计算更新,并且修改后的模型可以在高温下再现MP的点火延迟时间产生更好的预测。这项工作提供了MP单分子分解的全面研究,并可以作为了解更大的烷基酯热解的原型。








































 京公网安备 11010802027423号
京公网安备 11010802027423号