Letters in Drug Design & Discovery ( IF 1.2 ) Pub Date : 2020-07-31 , DOI: 10.2174/1570180817666200320105725 Yutao Zhao 1 , Xiaoqian Liu 1 , Jing Ouyang 1 , Yan Wang 1 , Shanyu Xu 2 , Dongdong Tian 1 , Hongzong Si 3
|
Background: In this study, modulators of human Chemotactic cytokine receptor 5 (CCR5) were described using a quantitative structure-activity relationship model (QSAR). This model was based on the molecule’s chemical structure.
Methods: All 56 compounds of CCR5 receptor antagonists were randomly separated into two sets, 43 were reserved for training and the other 13 for testing. In the course of this study, molecular models were drawn using ChemDraw software. By means of Hyperchem software as well as optimized with both AM1 (semi-empirical self-consistent-field molecular orbital) and MM+ (molecular mechanics plus force field), molecular models were described through numerous descriptors using CODESSA software.
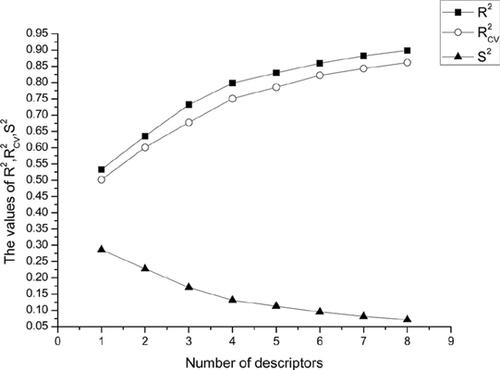
Results: Linear models were obtained by Heuristic Method (HM) software and nonlinear models were obtained using APS software with optimal descriptor combinations used to build linear QSAR models, involving a group of selected descriptors. As a result, values of the above two different sets were shown to result from 0.82 in testing and 0.86 in training in HM while 0.83 in testing and 0.88 in training in Gene Expression Programming (GEP).
Conclusion: From this method, the activity of molecules could be predicted, and the molecular structure could be changed to alter its IC50, avoiding the testing of large numbers of compounds.
中文翻译:
基于QSAR的代谢稳定的1-(3,3-二苯丙基)-哌啶基酰胺和尿素作为人类CCR5受体拮抗剂的IC50研究
背景:在这项研究中,使用定量结构-活性关系模型(QSAR)描述了人类趋化性细胞因子受体5(CCR5)的调节剂。该模型基于分子的化学结构。
方法:将56种CCR5受体拮抗剂化合物随机分为两组,保留43种用于训练,其余13种用于测试。在本研究过程中,使用ChemDraw软件绘制了分子模型。通过Hyperchem软件,并通过AM1(半经验自洽场分子轨道)和MM +(分子力学加力场)进行优化,使用CODESSA软件通过众多描述符描述了分子模型。
结果:使用启发式方法(HM)软件获得线性模型,使用APS软件获得非线性模型,并使用最佳描述符组合来构建线性QSAR模型,其中涉及一组选定的描述符。结果,显示上述两个不同集合的值来自HM中的测试0.82和0.86,而基因表达编程(GEP)的测试为0.83和0.88。
结论:通过该方法可以预测分子的活性,并可以改变分子结构以改变其IC50,从而避免了对大量化合物的测试。









































 京公网安备 11010802027423号
京公网安备 11010802027423号