Catalysis Today ( IF 5.2 ) Pub Date : 2020-07-30 , DOI: 10.1016/j.cattod.2020.07.039 Simuck F. Yuk 1 , Mal-Soon Lee 1 , Sneha A. Akhade 1, 2 , Manh-Thuong Nguyen 1 , Vassiliki-Alexandra Glezakou 1 , Roger Rousseau 1
|
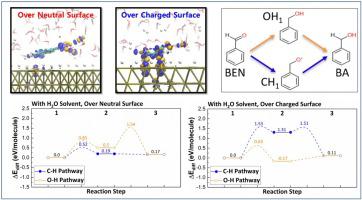
Understanding the hydrogenation of organic compounds in the aqueous phase has always been fundamentally important for improving carbon neutral pathways to fuels and value-added chemicals. In this study, we investigated both thermodynamic and kinetic profiles of benzaldehyde hydrogenation over the Pd(111) and Pt(111) metal surfaces using density functional theory (DFT) and ab initio molecular dynamic (AIMD) simulations. The adsorption of H2 shows the mixed preference of H adsorption sites on the Pt(111), while the fcc adsorption site is dominant for H on the Pd(111). When benzaldehyde is added to the systems, we observe a strong reduction of benzaldehyde on charged Pd (111) surface compared with that on neutral surface. In contrast, charged state of the Pt(111) surface does not change their interaction. Subsequent hydrogenation reaction of benzaldehyde over Pd(111), proceeding via Langmuir-Hinshelwood mechanism, is affected by two major factors: the presence of H2O solvent and surface charge. The presence of H2O solvent greatly reduces the activation energy of C H and OH bond formation during the hydrogenation process. Furthermore, the hydrogenation step via CH bond formation is preferred thermodynamically and kinetically over OH bond formation during thermocatalytic hydrogenation, while the opposite trend holds true during electrocatalytic hydrogenation.
H and OH bond formation during the hydrogenation process. Furthermore, the hydrogenation step via CH bond formation is preferred thermodynamically and kinetically over OH bond formation during thermocatalytic hydrogenation, while the opposite trend holds true during electrocatalytic hydrogenation.
中文翻译:
Pt族金属催化苯甲醛加氢的第一性原理研究
了解水相中有机化合物的加氢对于改善燃料和增值化学品的碳中和途径一直至关重要。在这项研究中,我们使用密度泛函理论 (DFT) 和从头算分子动力学 (AIMD) 模拟研究了苯甲醛加氢在 Pd(111) 和 Pt(111) 金属表面上的热力学和动力学曲线。H 2的吸附显示了 Pt(111) 上 H 吸附位点的混合偏好,而 fcc 吸附位点对 Pd(111) 上的 H 占主导地位。当将苯甲醛添加到系统中时,我们观察到与中性表面相比,带电 Pd (111) 表面的苯甲醛显着减少。相反,Pt(111) 表面的带电状态不会改变它们的相互作用。随后的苯甲醛在 Pd(111) 上的氢化反应,通过 Langmuir-Hinshelwood 机理进行,受两个主要因素的影响:H 2 O 溶剂的存在和表面电荷。H 2 O溶剂的存在大大降低了加氢过程中CH和OH键形成的活化能。此外,通过 C 的氢化步骤在热催化加氢过程中,H 键的形成在热力学和动力学上优于 O H 键的形成,而在电催化加氢过程中则相反。










































 京公网安备 11010802027423号
京公网安备 11010802027423号