当前位置:
X-MOL 学术
›
Hum. Mutat.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Expansion of the phenotypic spectrum of de novo missense variants in kinesin family member 1A (KIF1A).
Human Mutation ( IF 3.3 ) Pub Date : 2020-07-11 , DOI: 10.1002/humu.24079 Simranpreet Kaur 1, 2 , Nicole J Van Bergen 1, 2 , Kristen J Verhey 3 , Cameron J Nowell 4 , Breane Budaitis 5 , Yang Yue 3 , Carolyn Ellaway 6, 7 , Nicola Brunetti-Pierri 8, 9 , Gerarda Cappuccio 8, 9 , Irene Bruno 10 , Lia Boyle 11 , Vincenzo Nigro 10 , Annalaura Torella 10 , Tony Roscioli 12, 13 , Mark J Cowley 14, 15, 16 , Sean Massey 1 , Rhea Sonawane 17 , Matthew D Burton 18 , Bitten Schonewolf-Greulich 19 , Zeynep Tümer 19 , Wendy K Chung 20 , Wendy A Gold 21, 22, 23 , John Christodoulou 1, 2, 6, 24
Human Mutation ( IF 3.3 ) Pub Date : 2020-07-11 , DOI: 10.1002/humu.24079 Simranpreet Kaur 1, 2 , Nicole J Van Bergen 1, 2 , Kristen J Verhey 3 , Cameron J Nowell 4 , Breane Budaitis 5 , Yang Yue 3 , Carolyn Ellaway 6, 7 , Nicola Brunetti-Pierri 8, 9 , Gerarda Cappuccio 8, 9 , Irene Bruno 10 , Lia Boyle 11 , Vincenzo Nigro 10 , Annalaura Torella 10 , Tony Roscioli 12, 13 , Mark J Cowley 14, 15, 16 , Sean Massey 1 , Rhea Sonawane 17 , Matthew D Burton 18 , Bitten Schonewolf-Greulich 19 , Zeynep Tümer 19 , Wendy K Chung 20 , Wendy A Gold 21, 22, 23 , John Christodoulou 1, 2, 6, 24
Affiliation
|
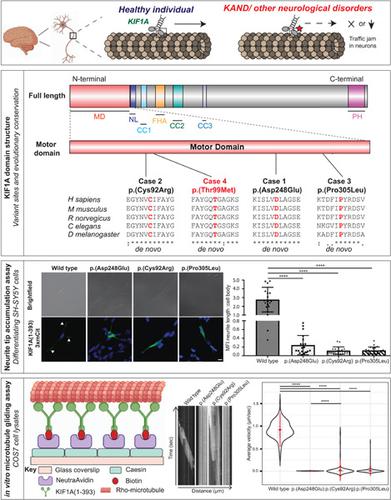
Defects in the motor domain of kinesin family member 1A (KIF1A), a neuron‐specific ATP‐dependent anterograde axonal transporter of synaptic cargo, are well‐recognized to cause a spectrum of neurological conditions, commonly known as KIF1A‐associated neurological disorders (KAND). Here, we report one mutation‐negative female with classic Rett syndrome (RTT) harboring a de novo heterozygous novel variant [NP_001230937.1:p.(Asp248Glu)] in the highly conserved motor domain of KIF1A. In addition, three individuals with severe neurodevelopmental disorder along with clinical features overlapping with KAND are also reported carrying de novo heterozygous novel [NP_001230937.1:p.(Cys92Arg) and p.(Pro305Leu)] or previously reported [NP_001230937.1:p.(Thr99Met)] variants in KIF1A. In silico tools predicted these variants to be likely pathogenic, and 3D molecular modeling predicted defective ATP hydrolysis and/or microtubule binding. Using the neurite tip accumulation assay, we demonstrated that all novel KIF1A variants significantly reduced the ability of the motor domain of KIF1A to accumulate along the neurite lengths of differentiated SH‐SY5Y cells. In vitro microtubule gliding assays showed significantly reduced velocities for the variant p.(Asp248Glu) and reduced microtubule binding for the p.(Cys92Arg) and p.(Pro305Leu) variants, suggesting a decreased ability of KIF1A to move along microtubules. Thus, this study further expanded the phenotypic characteristics of KAND individuals with pathogenic variants in the KIF1A motor domain to include clinical features commonly seen in RTT individuals.
中文翻译:

驱动蛋白家族成员 1A (KIF1A) 从头错义变异的表型谱扩展。
驱动蛋白家族成员 1A ( KIF1A ) 的运动域缺陷是一种神经元特异性 ATP 依赖的突触货物顺行轴突转运蛋白,被公认为会导致一系列神经系统疾病,通常称为KIF1A相关神经系统疾病 (KAND )。在这里,我们报告了一名患有经典 Rett 综合征 (RTT) 的突变阴性女性,在 KIF1A 的高度保守的运动结构域中含有一个新的杂合新变体 [NP_001230937.1:p.(Asp248Glu)]。此外,还报告了三名患有严重神经发育障碍以及与 KAND 重叠的临床特征的个体携带新杂合子 [NP_001230937.1:p.(Cys92Arg) 和 p.(Pro305Leu)] 或先前报道的 [NP_001230937.1:p .(Thr99Met)] KIF1A中的变体. 计算机工具预测这些变异可能是致病的,3D 分子建模预测有缺陷的 ATP 水解和/或微管结合。使用神经突尖端积累试验,我们证明了所有新的 KIF1A变体显着降低了 KIF1A 的运动结构域沿分化的 SH-SY5Y 细胞的神经突长度积累的能力。体外微管滑动试验显示变体 p.(Asp248Glu) 的速度显着降低,p.(Cys92Arg) 和 p.(Pro305Leu) 变体的微管结合减少,表明 KIF1A 沿微管移动的能力降低。因此,本研究进一步扩展了具有 KIF1A 运动域致病变异的 KAND 个体的表型特征,以包括 RTT 个体中常见的临床特征。
更新日期:2020-07-11
中文翻译:
驱动蛋白家族成员 1A (KIF1A) 从头错义变异的表型谱扩展。
驱动蛋白家族成员 1A ( KIF1A ) 的运动域缺陷是一种神经元特异性 ATP 依赖的突触货物顺行轴突转运蛋白,被公认为会导致一系列神经系统疾病,通常称为KIF1A相关神经系统疾病 (KAND )。在这里,我们报告了一名患有经典 Rett 综合征 (RTT) 的突变阴性女性,在 KIF1A 的高度保守的运动结构域中含有一个新的杂合新变体 [NP_001230937.1:p.(Asp248Glu)]。此外,还报告了三名患有严重神经发育障碍以及与 KAND 重叠的临床特征的个体携带新杂合子 [NP_001230937.1:p.(Cys92Arg) 和 p.(Pro305Leu)] 或先前报道的 [NP_001230937.1:p .(Thr99Met)] KIF1A中的变体. 计算机工具预测这些变异可能是致病的,3D 分子建模预测有缺陷的 ATP 水解和/或微管结合。使用神经突尖端积累试验,我们证明了所有新的 KIF1A变体显着降低了 KIF1A 的运动结构域沿分化的 SH-SY5Y 细胞的神经突长度积累的能力。体外微管滑动试验显示变体 p.(Asp248Glu) 的速度显着降低,p.(Cys92Arg) 和 p.(Pro305Leu) 变体的微管结合减少,表明 KIF1A 沿微管移动的能力降低。因此,本研究进一步扩展了具有 KIF1A 运动域致病变异的 KAND 个体的表型特征,以包括 RTT 个体中常见的临床特征。








































 京公网安备 11010802027423号
京公网安备 11010802027423号