Physica E: Low-dimensional Systems and Nanostructures ( IF 2.9 ) Pub Date : 2020-07-09 , DOI: 10.1016/j.physe.2020.114348 Huaguang Li , Feifei An , Jianxin Xie , Yan Wang
|
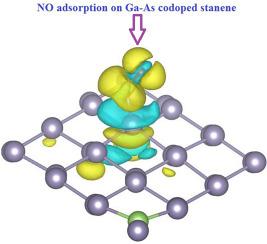
The density functional theory computations were performed to examine the interaction between NO molecule and Ga–As codoped stanene nanosheets. The band structure calculations indicated that the Ga–As codoped stanene acts a semiconductor material with a band gap around the Fermi level. Thus, the NO adsorption mechanism is inspected on the Ga–As codoped stanene systems. The results suggested that the NO binding to the stanene via the N center is stronger than that via the O center. Besides, the adsorption on the Ga–As codoped system was found to be more favorable in energy than that on the pure one. Therefore, Ga–As codoped system can provide most stable adsorption configurations with the adsorbed NO molecule. While the adsorption happens, the charge densities were mostly accumulated over the adsorbed NO molecule, which implies the acceptor behavior of the NO molecule in reaction with stanene nanosheets.
中文翻译:
DFT研究Ga和As对掺杂锡纳米片上NO气体分子的吸附行为
进行了密度泛函理论计算,以检查NO分子与Ga-As共掺杂锡纳米片之间的相互作用。能带结构计算表明,Ga-As共掺杂的锡充当半导体材料,其带隙在费米能级附近。因此,在Ga-As共掺杂锡系统上检查了NO的吸附机理。结果表明,NO通过N中心与锡的结合要强于通过O中心。此外,发现在Ga-As共掺杂体系上的吸附比纯体系上的吸附更有利。因此,Ga-As共掺杂体系可以为被吸附的NO分子提供最稳定的吸附构型。在发生吸附的同时,电荷密度大部分累积在吸附的NO分子上,









































 京公网安备 11010802027423号
京公网安备 11010802027423号