Journal of Molecular Graphics and Modelling ( IF 2.7 ) Pub Date : 2020-07-09 , DOI: 10.1016/j.jmgm.2020.107631 Shahnaz Ahmed 1 , Dhruba Jyoti Kalita 1
|
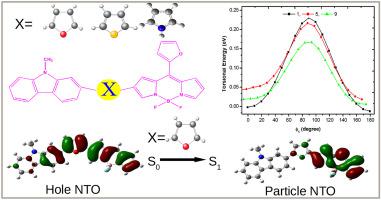
In this paper, we have examined the influence of torsional rigidity on the optoelectronic properties by imposing furan, pyrrole and thiophene unit on the BODIPY-carbazole based donor-acceptor systems employing density functional theory (DFT) formalism. We have designed 12 small conjugated molecules based on the donor (carbazole)-acceptor (BODIPY) approach using furan, pyrrole and thiophene unit as the bridging units. To study the torsional rigidity imparted by the bridging units we have performed potential energy surface (PES) analysis. Our study explores that among the bridging units furan and thiophene impart maximum and minimum rigidity on the systems respectively. Different parameters viz. distortion energy (), HOMO-LUMO gap ( values), ionization potential (IP), electron affinity (EA), bond length alteration (BLA) parameters, dipole moment values, reorganization energies for holes () and electrons (), electronic coupling matrix element (V), charge transfer rate (), hopping mobility (), radiative decay rate (kr) etc. have been calculated. The absorption and emission spectra of the BODIPY based compounds have been studied using TD-DFT. NTO analysis have also been performed for the dominant electronic transitions. Our calculations predict that compounds possessing pyrrole unit as the bridging unit and compounds in which BODIPY unit is meso substituted with pyrrole unit possesses greater amount of conjugation and as a result exhibit facile charge transport.
中文翻译:
在D-π-A方法的基础上合理设计BODIPY-咔唑类似物以进行便捷的电荷传输:DFT / TD-DFT研究。
在本文中,我们通过使用密度泛函理论(DFT)形式论将呋喃,吡咯和噻吩单元置于基于BODIPY-咔唑的供体-受体系统上,研究了扭转刚度对光电性能的影响。我们基于呋喃,吡咯和噻吩单元作为桥联单元,基于供体(咔唑)-受体(BODIPY)方法设计了12个小共轭分子。为了研究桥接单元的抗扭刚度,我们进行了势能面(PES)分析。我们的研究探索了呋喃和噻吩在桥接单元中分别赋予系统最大和最小的刚性。不同的参数 畸变能),HOMO-LUMO间隙( 值),电离势(IP),电子亲和力(EA),键长变化(BLA)参数,偶极矩值,空穴的重组能()和电子(),电子耦合矩阵元素(V),电荷转移率(),跳动性(),计算出辐射衰减率(k r)等。已经使用TD-DFT研究了BODIPY基化合物的吸收和发射光谱。还针对主要的电子跃迁进行了NTO分析。我们的计算预测,具有吡咯单元作为桥联单元的化合物以及BODIPY单元被吡咯单元取代的内消旋化合物具有更大的共轭量,因此显示出便捷的电荷传输。









































 京公网安备 11010802027423号
京公网安备 11010802027423号