当前位置:
X-MOL 学术
›
Clin. Genet.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
De novo missense variants in the RAP1B gene identified in two patients with syndromic thrombocytopenia.
Clinical Genetics ( IF 2.9 ) Pub Date : 2020-07-06 , DOI: 10.1111/cge.13807 Jan Hendrik Niemann 1 , Chen Du 1 , Susanne Morlot 1 , Gunnar Schmidt 1 , Bernd Auber 1 , Beate Kaune 1 , Gudrun Göhring 1 , Tim Ripperger 1 , Brigitte Schlegelberger 1 , Winfried Hofmann 1 , Thomas Smol 2 , Emilie Ait-Yahya 3 , Anna Raimbault 4 , Anne Lambilliotte 5 , Florence Petit 6 , Doris Steinemann 1
Clinical Genetics ( IF 2.9 ) Pub Date : 2020-07-06 , DOI: 10.1111/cge.13807 Jan Hendrik Niemann 1 , Chen Du 1 , Susanne Morlot 1 , Gunnar Schmidt 1 , Bernd Auber 1 , Beate Kaune 1 , Gudrun Göhring 1 , Tim Ripperger 1 , Brigitte Schlegelberger 1 , Winfried Hofmann 1 , Thomas Smol 2 , Emilie Ait-Yahya 3 , Anna Raimbault 4 , Anne Lambilliotte 5 , Florence Petit 6 , Doris Steinemann 1
Affiliation
|
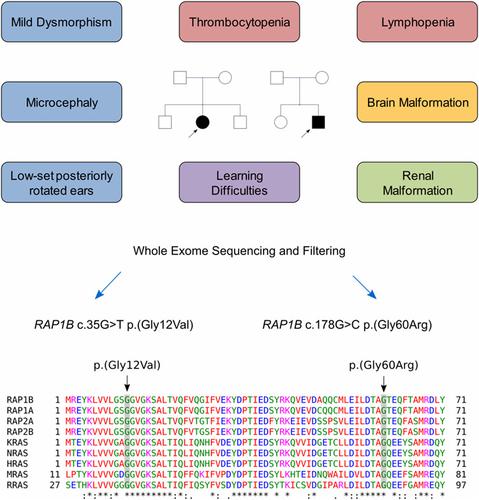
We present two independent cases of syndromic thrombocytopenia with multiple malformations, microcephaly, learning difficulties, dysmorphism and other features. Exome sequencing identified two novel de novo heterozygous variants in these patients, c.35G>T p.(Gly12Val) and c.178G>C p.(Gly60Arg), in the RAP1B gene (NM_001010942.2). These variants have not been described previously as germline variants, however functional studies in literature strongly suggest a clinical implication of these two activating hot spot positions. We hypothesize that pathogenic missense variants in the RAP1B gene cause congenital syndromic thrombocytopenia with a spectrum of associated malformations and dysmorphism, possibly through a gain of function mechanism.
中文翻译:

在两名患有血小板减少症的患者中发现了RAP1B基因的从头错义变异。
我们介绍了两个独立的综合征性血小板减少症,具有多种畸形,小头畸形,学习困难,畸形和其他特征。外显子组测序在RAP1B基因(NM_001010942.2)中确定了这些患者中的两个新的从头杂合变异,即c.35G> T p。(Gly12Val)和c.178G> C p。(Gly60Arg)。这些变体以前没有被描述为种系变体,但是文献中的功能研究强烈暗示了这两个激活热点位置的临床意义。我们假设RAP1B中的致病性错义变异 基因可能导致先天性综合征性血小板减少症,并伴有一系列相关的畸形和畸形,可能是通过功能机制获得的。
更新日期:2020-07-06
中文翻译:
在两名患有血小板减少症的患者中发现了RAP1B基因的从头错义变异。
我们介绍了两个独立的综合征性血小板减少症,具有多种畸形,小头畸形,学习困难,畸形和其他特征。外显子组测序在RAP1B基因(NM_001010942.2)中确定了这些患者中的两个新的从头杂合变异,即c.35G> T p。(Gly12Val)和c.178G> C p。(Gly60Arg)。这些变体以前没有被描述为种系变体,但是文献中的功能研究强烈暗示了这两个激活热点位置的临床意义。我们假设RAP1B中的致病性错义变异 基因可能导致先天性综合征性血小板减少症,并伴有一系列相关的畸形和畸形,可能是通过功能机制获得的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号