当前位置:
X-MOL 学术
›
Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Adsorption and diffusion of Au, Pt, and Co adatoms on SrTiO3(001) surfaces: A density functional theory study
Surface Science ( IF 2.1 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.susc.2020.121683 J. Buchwald , M. Hennes
Surface Science ( IF 2.1 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.susc.2020.121683 J. Buchwald , M. Hennes
|
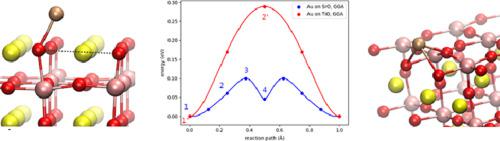
Abstract We use Density Functional Theory (DFT) and Hubbard model-based DFT + U calculations to determine preferential adsorption sites and energies of single, isolated Au, Pt, and Co atoms on planar SrTiO3(001) surfaces. Based on these results, we employ a nudged elastic band (NEB) approach to calculate relevant diffusion energy barriers for the three transition metal species on SrO and TiO2 terminated surfaces. This provides valuable quantitative input for future experimental and simulation studies and a sound basis to guide research aiming at controlling the microstructure of transition-metal-doped SrTiO3 thin films.
中文翻译:

Au、Pt 和 Co 吸附原子在 SrTiO3(001) 表面上的吸附和扩散:密度泛函理论研究
摘要 我们使用密度泛函理论 (DFT) 和基于哈伯德模型的 DFT + U 计算来确定平面 SrTiO3(001) 表面上单个孤立的 Au、Pt 和 Co 原子的优先吸附位点和能量。基于这些结果,我们采用轻推弹性带 (NEB) 方法来计算 SrO 和 TiO2 终止表面上三种过渡金属物种的相关扩散能垒。这为未来的实验和模拟研究提供了有价值的定量输入,并为指导旨在控制过渡金属掺杂 SrTiO3 薄膜微观结构的研究奠定了良好的基础。
更新日期:2020-11-01
中文翻译:
Au、Pt 和 Co 吸附原子在 SrTiO3(001) 表面上的吸附和扩散:密度泛函理论研究
摘要 我们使用密度泛函理论 (DFT) 和基于哈伯德模型的 DFT + U 计算来确定平面 SrTiO3(001) 表面上单个孤立的 Au、Pt 和 Co 原子的优先吸附位点和能量。基于这些结果,我们采用轻推弹性带 (NEB) 方法来计算 SrO 和 TiO2 终止表面上三种过渡金属物种的相关扩散能垒。这为未来的实验和模拟研究提供了有价值的定量输入,并为指导旨在控制过渡金属掺杂 SrTiO3 薄膜微观结构的研究奠定了良好的基础。









































 京公网安备 11010802027423号
京公网安备 11010802027423号