当前位置:
X-MOL 学术
›
Dalton Trans.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
d-orbital energy levels in planar [MIIF4]2-, [MII(NH3)4]2+ and [MII(CN)4]2- complexes: the nature of M-L π bonding and the implications for ligand field theory.
Dalton Transactions ( IF 3.5 ) Pub Date : 2020-07-03 , DOI: 10.1039/d0dt02022b Robert J Deeth 1
Dalton Transactions ( IF 3.5 ) Pub Date : 2020-07-03 , DOI: 10.1039/d0dt02022b Robert J Deeth 1
Affiliation
|
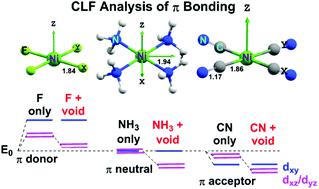
Qualitative MO theory predicts degenerate dπ orbitals for planar coordination complexes with formally σ-only ligands and the splitting energy, ΔEπ = E(dxy) − E(dxz,dyz), should be zero. For π-donor ligands, ΔEπ should be positive (dxy > dxz,yz) while for π-acceptors, ΔEπ should be negative (dxy < dxz,yz). However, experimental d–d spectra, ab initio ligand field theory (AI LFT) and crystal field theory for σ-only [M(NH3)4]2+ complexes give pronounced dπ splittings with ΔEπ around +2500 cm−1 for first-row, divalent metal ions. AI LFT further suggests ΔEπ values around +4500 cm−1 for [MF4]2− and +1000 cm−1 for [M(CN)4]2− species. The origins of these dπ orbital splittings can be traced to the effects of the ligand field potential surrounding the metal centre which includes not only the intrinsic metal–ligand π bonding but also substantial contributions from the ‘void’ regions above and below the molecular plane. The π component of the ‘void cell’ potentials increases ΔEπ which, if not explicitly taken into account, artificially enhances the apparent π-donor strength of the ligands. With the inclusion of void cell π interactions, even though the AI LFT d orbital sequence always places dxz,yz below dxy, the ligand field analysis provides a chemically-reasonable description of the M–L π interactions with cyanide being a weak π acceptor, ammonia being π-neutral and fluoride being a strong π donor. In the case of [Ni(CN)4]2−, ligand field calculations further show that, contrary to the recent claims of Oppenheim et al. (Inorg. Chem., 2019, 58, 15202) the sequence of the many-electron excited states is not a definitive guide to the underlying order of one-electron d orbital energies and that the observed sequence of nA2g > nEg > nB1g, n = 1 or 3, does not guarantee a d-orbital sequence of dxy < dxz,yz < dz2.
中文翻译:

平面[MIIF4] 2-,[MII(NH3)4] 2+和[MII(CN)4] 2-络合物中的d轨道能级:MLπ键的性质及其对配体场论的启示。
定性MO理论预测为平面配位络合物简并dπ轨道与正式σ-仅配体和分裂能量,Δ Ë π = È(d XY) - ë(d XZ,d YZ),应该是零。对于π供体配体,Δ È π应为正(d XY > d XZ,YZ),而π共受体,Δ È π应该是否定的(d XY <d XZ,YZ)。但是,实验性d–d光谱从头算配位场理论(AI LFT)和晶体场理论为σ-只[M(NH 3)4 ] 2+复合物给予显着的dπ分裂与Δ Ë π周围2500厘米-1为第一行,二价金属离子。AI LFT进一步表明Δ Ë π值周围4500厘米-1为[MF 4 ] 2-和千厘米-1对于[M(CN)4 ] 2-种类。这些dπ轨道分裂的起源可以追溯到金属中心周围的配体场电势的影响,其中不仅包括固有的金属-配体π键,还包括分子平面上方和下方的“空隙”区域的重要贡献。所述“无效细胞的潜能的π组分提高Δ ë π,如果不明确地考虑到,人为地提高了配体的表观π供体强度。通过包含空单元π相互作用,即使AI LFT d轨道序列始终将d xz,yz置于d xy之下,配体场分析提供了一种化学上合理的描述,其中氰化物为弱π受体,氨为π中性而氟化物为强π供体的M-Lπ相互作用。在[Ni(CN)4 ] 2−的情况下,配体场计算进一步表明,与Oppenheim等人的最新主张相反。(Inorg。化学式,2019年,58,15202)的多电子激发态的序列不是明确的指导单电子d轨道能量的基本顺序,并且所观察到的序列Ñ甲2克> Ñ Ë克> n B 1g,n= 1或3,不保证d xy <d xz,yz <d z 2的d轨道序列。
更新日期:2020-07-21
中文翻译:
平面[MIIF4] 2-,[MII(NH3)4] 2+和[MII(CN)4] 2-络合物中的d轨道能级:MLπ键的性质及其对配体场论的启示。
定性MO理论预测为平面配位络合物简并dπ轨道与正式σ-仅配体和分裂能量,Δ Ë π = È(d XY) - ë(d XZ,d YZ),应该是零。对于π供体配体,Δ È π应为正(d XY > d XZ,YZ),而π共受体,Δ È π应该是否定的(d XY <d XZ,YZ)。但是,实验性d–d光谱从头算配位场理论(AI LFT)和晶体场理论为σ-只[M(NH 3)4 ] 2+复合物给予显着的dπ分裂与Δ Ë π周围2500厘米-1为第一行,二价金属离子。AI LFT进一步表明Δ Ë π值周围4500厘米-1为[MF 4 ] 2-和千厘米-1对于[M(CN)4 ] 2-种类。这些dπ轨道分裂的起源可以追溯到金属中心周围的配体场电势的影响,其中不仅包括固有的金属-配体π键,还包括分子平面上方和下方的“空隙”区域的重要贡献。所述“无效细胞的潜能的π组分提高Δ ë π,如果不明确地考虑到,人为地提高了配体的表观π供体强度。通过包含空单元π相互作用,即使AI LFT d轨道序列始终将d xz,yz置于d xy之下,配体场分析提供了一种化学上合理的描述,其中氰化物为弱π受体,氨为π中性而氟化物为强π供体的M-Lπ相互作用。在[Ni(CN)4 ] 2−的情况下,配体场计算进一步表明,与Oppenheim等人的最新主张相反。(Inorg。化学式,2019年,58,15202)的多电子激发态的序列不是明确的指导单电子d轨道能量的基本顺序,并且所观察到的序列Ñ甲2克> Ñ Ë克> n B 1g,n= 1或3,不保证d xy <d xz,yz <d z 2的d轨道序列。










































 京公网安备 11010802027423号
京公网安备 11010802027423号