Computational and Theoretical Chemistry ( IF 3.0 ) Pub Date : 2020-07-03 , DOI: 10.1016/j.comptc.2020.112918 Jairo Castillo-Chará
|
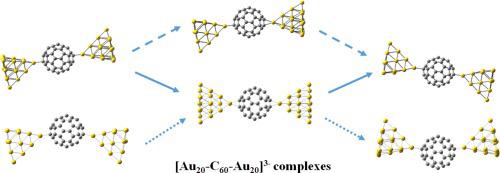
We have calculated molecular geometries, HOMO-LUMO gaps, Fermi levels and vibrational frequencies for the σ-linear, σ-bent and π-coordinated [Au20-C60-Au20]n− (n = 0–3) complexes using the density functional B3LYP method and LANL2DZ basis sets. Our calculations showed that the σ-bent η1(b)-[Au20-C60-Au20]3− complex is the global minimum structure with Au-C distances of 2.27 and 2.97 Å, respectively. The C60 calculated bouncing frequencies for the [Au20-C60-Au20]n− (n = 0–3) complexes appear at 18–60 cm−1 and compare well with the experimental value of 40 cm−1. The calculations of Mulliken charges indicate that the electrostatic interaction of the C60n− ions with the Au20 clusters plays a major role in the stability and electron-conducting properties of the [Au20-C60-Au20]n− (n = 0–3) complexes. The orbital analysis and calculated Fermi levels of the complexes indicate that the neutrally-charged singlet [Au20-C60-Au20] complexes have the HOMO-1 and LUMO+1 delocalized orbitals with Fermi levels at −5.18 eV that line up with the free C60 Fermi level.
中文翻译:
的分子性质的密度泛函计算[金20 -C 60 -Au 20 ] ñ -(N = 0,1,2,3)的复合物的模型
我们使用以下公式计算了σ线性,σ弯曲和π坐标[Au 20 -C 60 -Au 20 ] n −(n = 0–3)配合物的分子几何结构,HOMO-LUMO间隙,费米能级和振动频率。密度泛函B3LYP方法和LANL2DZ基集。我们的计算表明,σ弯曲η1 (b) -[Au 20- C 60- Au 20 ] 3-复合物是全局最小结构,Au-C距离分别为2.27和2.97Å。C 60为[Au 20 -C 60 -Au 20 ]计算的弹跳频率n −( n = 0–3)络合物出现在18–60 cm -1处,并与40 cm -1的实验值很好地比较。Mulliken电荷的计算表明,C 60 n-离子与Au 20团簇的静电相互作用在[Au 20 -C 60 -Au 20 ] n-(n = 0–3)复合体。配合物的轨道分析和计算的费米能级表明,中性带电单峰[Au 20 -C 60 -Au 20]配合物具有HOMO-1和LUMO + 1离域轨道,费米能级为-5.18 eV,与自由C 60费米能级一致。










































 京公网安备 11010802027423号
京公网安备 11010802027423号