当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Exfoliation Energy of Layered Materials by DFT-D: Beware of Dispersion!
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-07-01 , DOI: 10.1021/acs.jctc.0c00149 Michele Cutini 1 , Lorenzo Maschio 1 , Piero Ugliengo 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-07-01 , DOI: 10.1021/acs.jctc.0c00149 Michele Cutini 1 , Lorenzo Maschio 1 , Piero Ugliengo 1
Affiliation
|
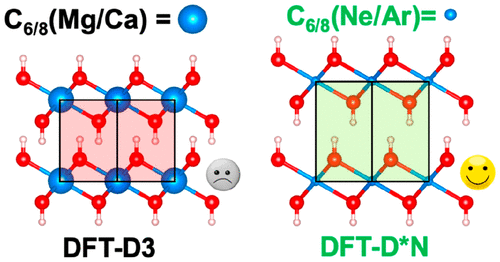
In this work, we have computed the exfoliation energy (the energy required to separate a single layer from the bulk structure), the interlayer distance, and the thermodynamic state functions for representative layered inorganic minerals such as Brucite, Portlandite, and Kaolinite, while leaving the more classical 2D transition-metal dichalcogenides (like MoS2) for future work. Such materials are interesting for several applications in the field of adsorption and in prebiotic chemistry. Their peculiar features are directly controlled by the exfoliation energy. In materials without cations/anions linking different layers, the interactions keeping the layers together are of weak nature, mainly dispersion London interactions and hydrogen bonds, somehow challenging to deal with computationally. We used Hartree–Fock (HF) and density functional theory (DFT) approaches focusing on the role of dispersion forces using the popular and widespread Grimme’s pairwise dispersion schemes (-D2 and -D3) and, as a reference method, the periodic MP2 approach based on localized orbitals (LMP2). The results are highly dependent on the choice of the scheme adopted to account for dispersion interactions. D2 and D3 schemes combined with either HF or DFT lead to overestimated exfoliation energies (about 2.5 and 1.7 times higher than LMP2 data for Brucite/Portlandite and Kaolinite) and underestimated interlayer distances (by about 3.5% for Brucite/Portlandite). The reason is that D2 and D3 corrections are based on neutral atomic parameters for each chemical element which, instead, behave as cations in the considered layered material (Mg, Ca, and Al), causing overattractive interaction between layers. More sophisticated dispersion corrections methods, like those based on nonlocal vdW functionals, many body dispersion model, and exchange-hole dipole moment not relying on atom-typing, are, in principle, better suited to describe the London interaction of ionic species. Nonetheless, we demonstrate that good results can be achieved also within the simpler D2 and D3 schemes, in agreement with previous literature suggestions, by adopting the dispersion coefficients of the preceding noble gas for the ionic species, leading to energetics in good agreement with LMP2 and structures closer to the experiments.
中文翻译:

DFT-D层状材料的剥落能量:当心分散!
在这项工作中,我们计算了剥落能量(将单层结构与块状结构分开所需的能量),层间距离以及代表性层状无机矿物(例如水镁石,硅酸盐和高岭石)的热力学状态函数,而保留了更经典的2D过渡金属二卤化金属(如MoS 2)以供将来工作。这样的材料对于吸附领域和益生元化学中的几种应用是令人感兴趣的。它们的独特特征直接由剥脱能量控制。在没有阳离子/阴离子连接不同层的材料中,将各层保持在一起的相互作用性质较弱,主要是分散伦敦相互作用和氢键,因此在计算上存在一定难度。我们使用Hartree–Fock(HF)和密度泛函理论(DFT)的方法,重点研究了使用流行和广泛使用的Grimme的成对色散方案(-D2和-D3)的色散力的作用,作为参考方法,使用了周期性MP2方法基于局部轨道(LMP2)。结果高度依赖于用于考虑色散相互作用的方案的选择。D2和D3方案与HF或DFT结合使用会导致高估剥落能量(对于水镁石/硅酸盐和高岭石,其剥落能分别比LMP2数据高约2.5和1.7倍),而层间距离则低估了(对于水镁石/硅酸盐,层间距离低了约3.5%)。原因是D2和D3校正基于每种化学元素的中性原子参数,而这些元素在所考虑的层状材料(Mg,Ca和Al)中表现为阳离子,从而导致层之间的吸引力太强。原则上,更复杂的色散校正方法(例如基于非局部vdW功能的校正,许多体色散模型和不依赖原子类型的交换孔偶极矩)更适合于描述离子物种的伦敦相互作用。尽管如此,
更新日期:2020-08-11
中文翻译:
DFT-D层状材料的剥落能量:当心分散!
在这项工作中,我们计算了剥落能量(将单层结构与块状结构分开所需的能量),层间距离以及代表性层状无机矿物(例如水镁石,硅酸盐和高岭石)的热力学状态函数,而保留了更经典的2D过渡金属二卤化金属(如MoS 2)以供将来工作。这样的材料对于吸附领域和益生元化学中的几种应用是令人感兴趣的。它们的独特特征直接由剥脱能量控制。在没有阳离子/阴离子连接不同层的材料中,将各层保持在一起的相互作用性质较弱,主要是分散伦敦相互作用和氢键,因此在计算上存在一定难度。我们使用Hartree–Fock(HF)和密度泛函理论(DFT)的方法,重点研究了使用流行和广泛使用的Grimme的成对色散方案(-D2和-D3)的色散力的作用,作为参考方法,使用了周期性MP2方法基于局部轨道(LMP2)。结果高度依赖于用于考虑色散相互作用的方案的选择。D2和D3方案与HF或DFT结合使用会导致高估剥落能量(对于水镁石/硅酸盐和高岭石,其剥落能分别比LMP2数据高约2.5和1.7倍),而层间距离则低估了(对于水镁石/硅酸盐,层间距离低了约3.5%)。原因是D2和D3校正基于每种化学元素的中性原子参数,而这些元素在所考虑的层状材料(Mg,Ca和Al)中表现为阳离子,从而导致层之间的吸引力太强。原则上,更复杂的色散校正方法(例如基于非局部vdW功能的校正,许多体色散模型和不依赖原子类型的交换孔偶极矩)更适合于描述离子物种的伦敦相互作用。尽管如此,









































 京公网安备 11010802027423号
京公网安备 11010802027423号