当前位置:
X-MOL 学术
›
ChemistrySelect
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Designing Triphenylamine‐Configured Donor Materials with Promising Photovoltaic Properties for Highly Efficient Organic Solar Cells
ChemistrySelect ( IF 1.9 ) Pub Date : 2020-07-01 , DOI: 10.1002/slct.202001989 Muhammad Bilal Ahmed Siddique 1 , Riaz Hussain 2 , Sabir Ali Siddique 3 , Muhammad Yasir Mehboob 2 , Zobia Irshad 4 , Javed Iqbal 5 , Muhammad Adnan 4
ChemistrySelect ( IF 1.9 ) Pub Date : 2020-07-01 , DOI: 10.1002/slct.202001989 Muhammad Bilal Ahmed Siddique 1 , Riaz Hussain 2 , Sabir Ali Siddique 3 , Muhammad Yasir Mehboob 2 , Zobia Irshad 4 , Javed Iqbal 5 , Muhammad Adnan 4
Affiliation
|
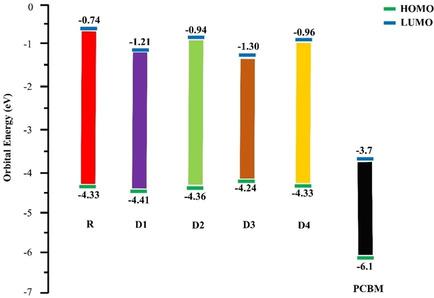
The increasing demand for energy expedited the development of efficient photovoltaic materials. Herein, four triphenylamine based push pull donor materials (D1‐D4 ) have been designed. The optical, electronic, photophysical properties and excited state energy of D1‐D4 have been investigated theoretically through DFT calculations at B3LYP/6‐31G (d,p) level of theory and compared with reference molecule R . The theoretical study of the designed molecules (D1 ‐D4 ) and reference molecule R with TD−B3LYP/6‐31G (d,p) level of theory was carried out both in gaseous and solvent (Chloroform/CPCM) phase to investigate their excited state properties. All the designed molecules D1 ‐D4 exhibited broad and intense absorption peaks in the visible spectrum from 300 nm to 450 nm range with narrow HOMO‐LUMO energy gaps as compared to reference R . The dipole moment of designed molecules D1 ‐D4 are higher than reference molecule R in both gas and solvent phase which may help to enhance the photovoltaic stability of organic solar cells devices. The open‐circuit voltages  of designed molecules, D1‐D4 and the reference molecule R compared to PCBM are 0.71 V, 0.66 V, 0.54 V, 0.63 V, and 0.63 V, respectively. The % ETC for designed molecules insolvent as well as in the gas phase is lower than the reference molecule R which enables them to excite rapidly both in gas and solvent phase respectively. The hole and electron transfer mobilities values indicate that the designed molecules have a better electron and hole transport mobility values as compared to reference molecule R . Furthermore, conceptualized molecules are better and thus are recommended to experimentalists for out‐looking future developments of solar cells.
of designed molecules, D1‐D4 and the reference molecule R compared to PCBM are 0.71 V, 0.66 V, 0.54 V, 0.63 V, and 0.63 V, respectively. The % ETC for designed molecules insolvent as well as in the gas phase is lower than the reference molecule R which enables them to excite rapidly both in gas and solvent phase respectively. The hole and electron transfer mobilities values indicate that the designed molecules have a better electron and hole transport mobility values as compared to reference molecule R . Furthermore, conceptualized molecules are better and thus are recommended to experimentalists for out‐looking future developments of solar cells.
中文翻译:

设计具有优异光伏性能的三苯胺配置的供体材料,用于高效有机太阳能电池
能源需求的增长加速了高效光伏材料的开发。在此,已经设计了四种基于三苯胺的推拉供体材料(D1-D4)。D1-D4的光学,电子,光物理性质和激发态能已在理论上以B3LYP / 6-31G(d,p)水平通过DFT计算进行了研究,并与参考分子R进行了比较。在气相和溶剂相(氯仿/ CPCM)中对设计分子(D1 - D4)和参考分子R的理论水平为TD-B3LYP / 6-31G(d,p)进行了理论研究。状态属性。所有设计的分子与参考R相比,D1 - D4在300 nm至450 nm范围的可见光谱中显示出宽而强烈的吸收峰,并且HOMO-LUMO能隙较窄。在气相和溶剂相中,设计分子D1 - D4的偶极矩均高于参考分子R,这可能有助于增强有机太阳能电池装置的光伏稳定性。设计分子D1-D4和参考分子R的开路电压与PCBM相比,分别为0.71 V,0.66 V,0.54 V,0.63 V和0.63 V. 设计的无溶剂分子以及在气相中的%ETC低于参考分子R,这使它们分别在气相和溶剂相中都能迅速激发。空穴和电子转移迁移率值表明,与参考分子R相比,设计的分子具有更好的电子和空穴迁移率值。此外,概念化的分子更好,因此建议实验者忽略太阳能电池的未来发展。
更新日期:2020-07-01
of designed molecules, D1‐D4 and the reference molecule R compared to PCBM are 0.71 V, 0.66 V, 0.54 V, 0.63 V, and 0.63 V, respectively. The % ETC for designed molecules insolvent as well as in the gas phase is lower than the reference molecule R which enables them to excite rapidly both in gas and solvent phase respectively. The hole and electron transfer mobilities values indicate that the designed molecules have a better electron and hole transport mobility values as compared to reference molecule R . Furthermore, conceptualized molecules are better and thus are recommended to experimentalists for out‐looking future developments of solar cells.
中文翻译:
设计具有优异光伏性能的三苯胺配置的供体材料,用于高效有机太阳能电池
能源需求的增长加速了高效光伏材料的开发。在此,已经设计了四种基于三苯胺的推拉供体材料(D1-D4)。D1-D4的光学,电子,光物理性质和激发态能已在理论上以B3LYP / 6-31G(d,p)水平通过DFT计算进行了研究,并与参考分子R进行了比较。在气相和溶剂相(氯仿/ CPCM)中对设计分子(D1 - D4)和参考分子R的理论水平为TD-B3LYP / 6-31G(d,p)进行了理论研究。状态属性。所有设计的分子与参考R相比,D1 - D4在300 nm至450 nm范围的可见光谱中显示出宽而强烈的吸收峰,并且HOMO-LUMO能隙较窄。在气相和溶剂相中,设计分子D1 - D4的偶极矩均高于参考分子R,这可能有助于增强有机太阳能电池装置的光伏稳定性。设计分子D1-D4和参考分子R的开路电压
与PCBM相比,分别为0.71 V,0.66 V,0.54 V,0.63 V和0.63 V. 设计的无溶剂分子以及在气相中的%ETC低于参考分子R,这使它们分别在气相和溶剂相中都能迅速激发。空穴和电子转移迁移率值表明,与参考分子R相比,设计的分子具有更好的电子和空穴迁移率值。此外,概念化的分子更好,因此建议实验者忽略太阳能电池的未来发展。










































 京公网安备 11010802027423号
京公网安备 11010802027423号