当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Trajectory Surface Hopping Nonadiabatic Molecular Dynamics with Kohn-Sham ΔSCF for Condensed-Phase Systems.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-06-30 , DOI: 10.1021/acs.jctc.0c00372 Momir Mališ 1 , Sandra Luber 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-06-30 , DOI: 10.1021/acs.jctc.0c00372 Momir Mališ 1 , Sandra Luber 1
Affiliation
|
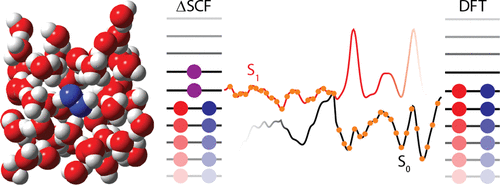
We present an efficient approach for surface hopping-based nonadiabatic dynamics in the condensed phase. For the systems studied, a restricted Kohn–Sham orbital formulation of the delta self-consistent field (ΔSCF) method was used for efficient calculation of excited electronic states. Time-dependent density functional theory (DFT) is applied to aid excited-state SCF convergence and provide guess electronic state densities. Aside from that the Landau–Zener procedure simplifies the surface hopping between electronic states. By utilizing the combined Gaussian and plane waves approach with periodic boundary conditions the method is easily applicable to full atomistic DFT simulations of condensed-phase systems and was used to study the nonradiative deactivation mechanism of photoexcited diimide in water solution.
中文翻译:

凝聚相系统中具有Kohn-ShamΔSCF的轨迹跳变非绝热分子动力学。
我们为凝结阶段基于表面跳跃的非绝热动力学提出了一种有效的方法。对于所研究的系统,使用三角洲自洽场(ΔSCF)方法的受限Kohn-Sham轨道公式来有效地计算激发电子态。随时间变化的密度泛函理论(DFT)用于辅助激发态SCF收敛并提供猜测的电子态密度。除此之外,Landau–Zener程序还简化了电子状态之间的表面跳变。通过利用具有周期性边界条件的高斯和平面波相结合的方法,该方法很容易适用于凝聚相系统的全原子DFT模拟,并用于研究光激发二酰亚胺在水溶液中的非辐射失活机理。
更新日期:2020-07-14
中文翻译:
凝聚相系统中具有Kohn-ShamΔSCF的轨迹跳变非绝热分子动力学。
我们为凝结阶段基于表面跳跃的非绝热动力学提出了一种有效的方法。对于所研究的系统,使用三角洲自洽场(ΔSCF)方法的受限Kohn-Sham轨道公式来有效地计算激发电子态。随时间变化的密度泛函理论(DFT)用于辅助激发态SCF收敛并提供猜测的电子态密度。除此之外,Landau–Zener程序还简化了电子状态之间的表面跳变。通过利用具有周期性边界条件的高斯和平面波相结合的方法,该方法很容易适用于凝聚相系统的全原子DFT模拟,并用于研究光激发二酰亚胺在水溶液中的非辐射失活机理。










































 京公网安备 11010802027423号
京公网安备 11010802027423号