当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
General Protocol for the Accurate Prediction of Molecular 13C/1H NMR Chemical Shifts via Machine Learning Augmented DFT.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-06-30 , DOI: 10.1021/acs.jcim.0c00388 Peng Gao 1 , Jun Zhang 2 , Qian Peng 3 , Jie Zhang 4 , Vassiliki-Alexandra Glezakou 2
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-06-30 , DOI: 10.1021/acs.jcim.0c00388 Peng Gao 1 , Jun Zhang 2 , Qian Peng 3 , Jie Zhang 4 , Vassiliki-Alexandra Glezakou 2
Affiliation
|
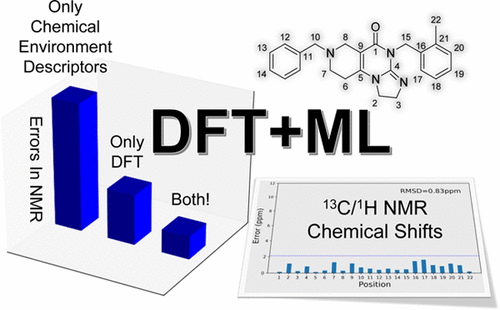
An accurate prediction of NMR chemical shifts at affordable computational cost is very important for different types of structural assignments in experimental studies. Density functional theory (DFT) and gauge-including atomic orbital (GIAO) are two of the most popular computational methods for NMR calculation, yet they often fail to resolve ambiguities in structural assignments. Here, we present a new method that uses machine learning (ML) techniques (DFT + ML) that significantly increases the accuracy of 13C/1H NMR chemical shift prediction for a variety of organic molecules. The input of the generalizable DFT + ML model contains two critical parts: one is a vector providing insights into chemical environments, which can be evaluated without knowing the exact geometry of the molecule; the other one is the DFT-calculated isotropic shielding constant. The DFT + ML model was trained with a data set containing 476 13C and 270 1H experimental chemical shifts. For the DFT methods used here, the root mean square deviations (RMSDs) for the errors between predicted and experimental 13C/1H chemical shifts can be as small as 2.10/0.18 ppm, which is much lower than those from simple DFT (5.54/0.25 ppm), or DFT + linear regression (LR) (4.77/0.23 ppm) approaches. It also has a smaller maximum absolute error than two previously proposed NMR-predicting ML models. The robustness of the DFT + ML model is tested on two classes of organic molecules (TIC10 and hyacinthacines), where the correct isomers were unambiguously assigned to the experimental ones. Overall, the DFT + ML model shows promise for structural assignments in a variety of systems, including stereoisomers, that are often challenging to determine experimentally.
中文翻译:

通过机器学习增强DFT准确预测分子13C / 1H NMR化学位移的通用协议。
以可承受的计算成本准确预测NMR化学位移对于实验研究中不同类型的结构分配非常重要。密度泛函理论(DFT)和包括轨距的原子轨(GIAO)是NMR计算中最流行的两种计算方法,但它们通常无法解决结构分配中的歧义。在这里,我们提出了一种使用机器学习(ML)技术(DFT + ML)的新方法,该方法显着提高了13 C / 1的精度各种有机分子的1 H NMR化学位移预测。泛化DFT + ML模型的输入包含两个关键部分:一个是提供对化学环境的洞察力的向量,可以在不知道分子确切几何形状的情况下进行评估;另一个是DFT计算的各向同性屏蔽常数。使用包含476 13 C和270 1 H实验化学位移的数据集训练了DFT + ML模型。对于此处使用的DFT方法,预测的和实验的13 C / 1之间的误差的均方根偏差(RMSD)H化学位移可以小到2.10 / 0.18 ppm,远低于简单DFT(5.54 / 0.25 ppm)或DFT +线性回归(LR)方法(4.77 / 0.23 ppm)的化学位移。它也具有比两个以前提出的NMR预测ML模型小的最大绝对误差。DFT + ML模型的稳健性在两类有机分子(TIC10和扁豆碱)上进行了测试,其中正确的异构体明确分配给了实验分子。总体而言,DFT + ML模型显示了在包括立体异构体在内的各种系统中进行结构分配的希望,这在实验上往往难以确定。
更新日期:2020-08-24
中文翻译:
通过机器学习增强DFT准确预测分子13C / 1H NMR化学位移的通用协议。
以可承受的计算成本准确预测NMR化学位移对于实验研究中不同类型的结构分配非常重要。密度泛函理论(DFT)和包括轨距的原子轨(GIAO)是NMR计算中最流行的两种计算方法,但它们通常无法解决结构分配中的歧义。在这里,我们提出了一种使用机器学习(ML)技术(DFT + ML)的新方法,该方法显着提高了13 C / 1的精度各种有机分子的1 H NMR化学位移预测。泛化DFT + ML模型的输入包含两个关键部分:一个是提供对化学环境的洞察力的向量,可以在不知道分子确切几何形状的情况下进行评估;另一个是DFT计算的各向同性屏蔽常数。使用包含476 13 C和270 1 H实验化学位移的数据集训练了DFT + ML模型。对于此处使用的DFT方法,预测的和实验的13 C / 1之间的误差的均方根偏差(RMSD)H化学位移可以小到2.10 / 0.18 ppm,远低于简单DFT(5.54 / 0.25 ppm)或DFT +线性回归(LR)方法(4.77 / 0.23 ppm)的化学位移。它也具有比两个以前提出的NMR预测ML模型小的最大绝对误差。DFT + ML模型的稳健性在两类有机分子(TIC10和扁豆碱)上进行了测试,其中正确的异构体明确分配给了实验分子。总体而言,DFT + ML模型显示了在包括立体异构体在内的各种系统中进行结构分配的希望,这在实验上往往难以确定。










































 京公网安备 11010802027423号
京公网安备 11010802027423号