Journal of Molecular Biology ( IF 4.7 ) Pub Date : 2020-06-27 , DOI: 10.1016/j.jmb.2020.06.021 Jennifer C Boatz 1 , Talia Piretra 1 , Alessia Lasorsa 2 , Irina Matlahov 3 , James F Conway 1 , Patrick C A van der Wel 3
|
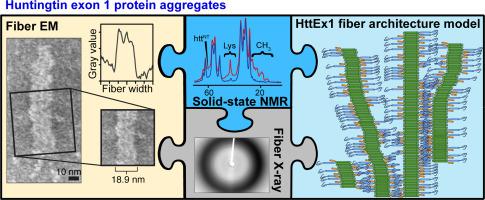
Huntington's disease is a progressive neurodegenerative disease caused by expansion of the polyglutamine domain in the first exon of huntingtin (HttEx1). The extent of expansion correlates with disease progression and formation of amyloid-like protein deposits within the brain. The latter display polymorphism at the microscopic level, both in cerebral tissue and in vitro. Such polymorphism can dramatically influence cytotoxicity, leading to much interest in the conditions and mechanisms that dictate the formation of polymorphs. We examine conditions that govern HttEx1 polymorphism in vitro, including concentration and the role of the non-polyglutamine flanking domains. Using electron microscopy, we observe polymorphs that differ in width and tendency for higher-order bundling. Strikingly, aggregation yields different polymorphs at low and high concentrations. Narrow filaments dominate at low concentrations that may be more relevant in vivo. We dissect the role of N- and C-terminal flanking domains using protein with the former (httNT or N17) largely removed. The truncated protein is generated by trypsin cleavage of soluble HttEx1 fusion protein, which we analyze in some detail. Dye binding and solid-state NMR studies reveal changes in fibril surface characteristics and flanking domain mobility. Higher-order interactions appear facilitated by the C-terminal tail, while the polyglutamine forms an amyloid core resembling those of other polyglutamine deposits. Fibril-surface-mediated branching, previously attributed to secondary nucleation, is reduced in absence of httNT. A new model for the architecture of the HttEx1 filaments is presented and discussed in context of the assembly mechanism and biological activity.
中文翻译:
聚集突变亨廷顿外显子1的原丝结构和超分子多态性。
亨廷顿舞蹈病是由亨廷顿蛋白第一个外显子(HttEx1)中的聚谷氨酰胺结构域扩展引起的进行性神经退行性疾病。扩展的程度与疾病的进展以及脑内淀粉样蛋白沉积物的形成有关。后者在脑组织和体外均在微观水平上表现出多态性。这种多态性可以极大地影响细胞毒性,导致人们对决定多态性形成的条件和机制产生了极大的兴趣。我们研究了在体外控制HttEx1多态性的条件,包括非多聚谷氨酰胺侧翼结构域的浓度和作用。使用电子显微镜,我们观察到宽度和高阶束缚趋势不同的多晶型物。令人惊讶的是,聚集在低浓度和高浓度下会产生不同的多晶型物。低浓度的细丝占主导地位,这在体内可能更重要。我们使用前者(htt NT)来分析N和C末端侧翼结构域的作用或N17)在很大程度上删除了 截短的蛋白是由胰蛋白酶裂解可溶性HttEx1融合蛋白产生的,我们对此进行了详细分析。染料结合和固态NMR研究揭示了原纤维表面特征和侧链结构域迁移率的变化。C端尾部促进了更高阶的相互作用,而聚谷氨酰胺形成了与其他聚谷氨酰胺沉积物相似的淀粉样核心。在没有HTT NT的情况下,原先归因于二次成核的原纤维表面介导的分支减少。在装配机理和生物活性的背景下,提出并讨论了HttEx1细丝结构的新模型。










































 京公网安备 11010802027423号
京公网安备 11010802027423号