当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
First-principles prediction of silicon nanocones: Stability and electronic properties
Computational Materials Science ( IF 3.3 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.commatsci.2020.109885 A. Freitas , C.G. Bezerra , L.D. Machado , S. Azevedo
Computational Materials Science ( IF 3.3 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.commatsci.2020.109885 A. Freitas , C.G. Bezerra , L.D. Machado , S. Azevedo
|
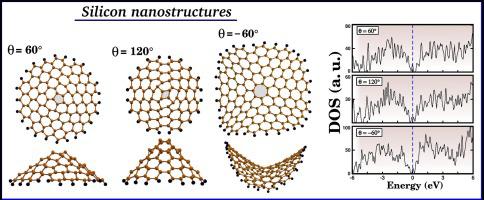
Abstract Despite the advanced stage of studies on carbon and BN nanocones, with important applications in nanotechnology, systematic investigations on nanocones composed of other atom types are still lacking. Here, we combine density functional theory (DFT) and ab initio molecular dynamics (AIMD) simulations to study the stability and electronic properties of silicon nanocones with different disclination angles ( θ = 60 ∘ , 120 ∘ , and 180 ∘ ). These structures exhibit structural deformation at the edge and apex regions, and also present the same puckered form of silicene. We find that the minimum energy structure is the nanocone with a disclination angle of 60 ∘ . Moreover, our ab initio molecular dynamics simulations reveal that this nanocone should remain stable at T = 300 K. We find that all investigated nanocones are metallic and exhibit an energy level located exactly at the Fermi energy. Regarding the magnetic properties, all investigated structures present unpaired electrons, which induce a total spin of 1 / 2 or 1. Additionally, we demonstrate that the substitutional doping of silicon nanocones with P and N atoms is energetically favorable, and also promotes the opening of the band gap. Our calculations add a new class of nanostructures to the increasing library of silicon materials.
中文翻译:

硅纳米锥的第一性原理预测:稳定性和电子特性
摘要 尽管碳和BN纳米锥的研究已进入先进阶段,在纳米技术中具有重要应用,但对其他原子类型组成的纳米锥的系统研究仍然缺乏。在这里,我们结合密度泛函理论 (DFT) 和 ab initio 分子动力学 (AIMD) 模拟来研究具有不同向错角( θ = 60 ∘ 、120 ∘ 和 180 ∘ )的硅纳米锥的稳定性和电子特性。这些结构在边缘和顶点区域表现出结构变形,并且还呈现出相同的硅烯褶皱形式。我们发现最小能量结构是向错角为 60 ∘ 的纳米锥。此外,我们的 ab initio 分子动力学模拟表明,该纳米锥应在 T = 300 K 时保持稳定。我们发现所有研究的纳米锥都是金属的,并且表现出的能级恰好位于费米能级。关于磁性,所有研究的结构都存在不成对的电子,导致总自旋为 1 / 2 或 1。此外,我们证明了硅纳米锥与 P 和 N 原子的置换掺杂在能量上是有利的,并且还促进了带隙。我们的计算为不断增加的硅材料库添加了一类新的纳米结构。并且也促进了带隙的打开。我们的计算为不断增加的硅材料库添加了一类新的纳米结构。并且也促进了带隙的打开。我们的计算为不断增加的硅材料库添加了一类新的纳米结构。
更新日期:2020-11-01
中文翻译:
硅纳米锥的第一性原理预测:稳定性和电子特性
摘要 尽管碳和BN纳米锥的研究已进入先进阶段,在纳米技术中具有重要应用,但对其他原子类型组成的纳米锥的系统研究仍然缺乏。在这里,我们结合密度泛函理论 (DFT) 和 ab initio 分子动力学 (AIMD) 模拟来研究具有不同向错角( θ = 60 ∘ 、120 ∘ 和 180 ∘ )的硅纳米锥的稳定性和电子特性。这些结构在边缘和顶点区域表现出结构变形,并且还呈现出相同的硅烯褶皱形式。我们发现最小能量结构是向错角为 60 ∘ 的纳米锥。此外,我们的 ab initio 分子动力学模拟表明,该纳米锥应在 T = 300 K 时保持稳定。我们发现所有研究的纳米锥都是金属的,并且表现出的能级恰好位于费米能级。关于磁性,所有研究的结构都存在不成对的电子,导致总自旋为 1 / 2 或 1。此外,我们证明了硅纳米锥与 P 和 N 原子的置换掺杂在能量上是有利的,并且还促进了带隙。我们的计算为不断增加的硅材料库添加了一类新的纳米结构。并且也促进了带隙的打开。我们的计算为不断增加的硅材料库添加了一类新的纳米结构。并且也促进了带隙的打开。我们的计算为不断增加的硅材料库添加了一类新的纳米结构。


























 京公网安备 11010802027423号
京公网安备 11010802027423号