当前位置:
X-MOL 学术
›
Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational study of the adsorption of bimetallic clusters on alumina substrate
Surface Science ( IF 2.1 ) Pub Date : 2020-10-01 , DOI: 10.1016/j.susc.2020.121682 Nusaiba Zaman , Karima Lasri , Kah Chun Lau , Khalil Amine , Abdelkader Kara
Surface Science ( IF 2.1 ) Pub Date : 2020-10-01 , DOI: 10.1016/j.susc.2020.121682 Nusaiba Zaman , Karima Lasri , Kah Chun Lau , Khalil Amine , Abdelkader Kara
|
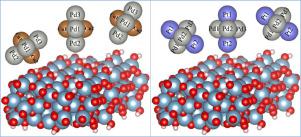
Abstract We performed computational investigation of the adsorption of bimetallic Pd3M2 (where M changes from Ag, Au, Co, Cu, Mn, Ni, Pt, and Ru) cluster on the hydroxylated alumina surface. Previously, it was shown that small silver cluster can control the rate of discharge at the cathode of lithium-oxygen battery. The gap near the fermi energy was shown to control the oxygen reduction, an important reaction for LiO2 formation. Controlling the gap would ultimately control the rate of LiO2 formation. One can vary the size of the cluster to vary the gap, however, this “knob” provides limited variance in the gap. Alloying, in combination with size variation offer a much wider control of the gap, hence the LiO2 formation. Using Density Functional Theory (DFT), we determined the most stable geometry of the bimetallic clusters Pd3M2 and calculated the binding energies of these clusters on the alumina substrate which ranges from 0.2 eV to 0.25 eV depending upon the composition of the alloy-cluster, its orientation and the adsorption site. We also find that Pd atoms bind strongly with the substrate oxygen atoms with an average short bond-length of about 2.2 A. We explored how the gap at the Fermi level of the system varies as a function of elemental composition and the calculated gap ranges from 0 meV to 90 meV. Charges distribution using Bader analysis was also performed to probe how charges are transferred between the cluster and the substrate. These preliminary results will open the door for more systematic studies of alloy clusters of different size and stoichiometry for Li-O2 battery cathode design.
中文翻译:

双金属团簇在氧化铝基底上吸附的计算研究
摘要 我们对双金属 Pd3M2(其中 M 从 Ag、Au、Co、Cu、Mn、Ni、Pt 和 Ru 变化)簇在羟基化氧化铝表面上的吸附进行了计算研究。之前的研究表明,小银簇可以控制锂氧电池阴极的放电速率。费米能量附近的间隙被证明可以控制氧还原,这是形成 LiO2 的重要反应。控制间隙将最终控制 LiO2 的形成速率。可以改变集群的大小来改变间隙,但是,这个“旋钮”提供的间隙变化有限。合金化与尺寸变化相结合,可以更广泛地控制间隙,从而形成 LiO2。使用密度泛函理论(DFT),我们确定了双金属团簇 Pd3M2 的最稳定几何形状,并计算了这些团簇在氧化铝基板上的结合能,其范围为 0.2 eV 至 0.25 eV,具体取决于合金团簇的组成、其取向和吸附位点。我们还发现 Pd 原子与底物氧原子强烈结合,平均短键长约为 2.2 A。我们探索了系统费米能级的间隙如何作为元素组成的函数而变化,计算的间隙范围为0 meV 到 90 meV。还进行了使用 Bader 分析的电荷分布,以探测电荷如何在簇和基板之间转移。这些初步结果将为更系统地研究不同尺寸和化学计量的 Li-O2 电池正极设计合金簇打开大门。
更新日期:2020-10-01
中文翻译:
双金属团簇在氧化铝基底上吸附的计算研究
摘要 我们对双金属 Pd3M2(其中 M 从 Ag、Au、Co、Cu、Mn、Ni、Pt 和 Ru 变化)簇在羟基化氧化铝表面上的吸附进行了计算研究。之前的研究表明,小银簇可以控制锂氧电池阴极的放电速率。费米能量附近的间隙被证明可以控制氧还原,这是形成 LiO2 的重要反应。控制间隙将最终控制 LiO2 的形成速率。可以改变集群的大小来改变间隙,但是,这个“旋钮”提供的间隙变化有限。合金化与尺寸变化相结合,可以更广泛地控制间隙,从而形成 LiO2。使用密度泛函理论(DFT),我们确定了双金属团簇 Pd3M2 的最稳定几何形状,并计算了这些团簇在氧化铝基板上的结合能,其范围为 0.2 eV 至 0.25 eV,具体取决于合金团簇的组成、其取向和吸附位点。我们还发现 Pd 原子与底物氧原子强烈结合,平均短键长约为 2.2 A。我们探索了系统费米能级的间隙如何作为元素组成的函数而变化,计算的间隙范围为0 meV 到 90 meV。还进行了使用 Bader 分析的电荷分布,以探测电荷如何在簇和基板之间转移。这些初步结果将为更系统地研究不同尺寸和化学计量的 Li-O2 电池正极设计合金簇打开大门。










































 京公网安备 11010802027423号
京公网安备 11010802027423号