当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Improving Proteoform Identifications in Complex Systems Through Integration of Bottom-Up and Top-Down Data.
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2020-06-25 , DOI: 10.1021/acs.jproteome.0c00332 Leah V Schaffer 1 , Robert J Millikin 1 , Michael R Shortreed 1 , Mark Scalf 1 , Lloyd M Smith 1
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2020-06-25 , DOI: 10.1021/acs.jproteome.0c00332 Leah V Schaffer 1 , Robert J Millikin 1 , Michael R Shortreed 1 , Mark Scalf 1 , Lloyd M Smith 1
Affiliation
|
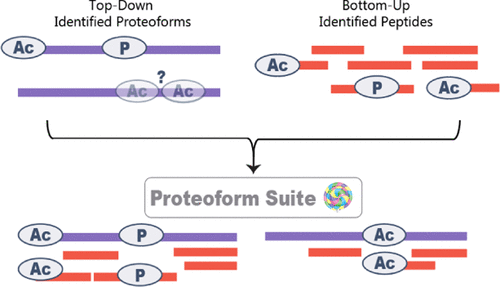
Cellular functions are performed by a vast and diverse set of proteoforms. Proteoforms are the specific forms of proteins produced as a result of genetic variations, RNA splicing, and post-translational modifications (PTMs). Top-down mass spectrometric analysis of intact proteins enables proteoform identification, including proteoforms derived from sequence cleavage events or harboring multiple PTMs. In contrast, bottom-up proteomics identifies peptides, which necessitates protein inference and does not yield proteoform identifications. We seek here to exploit the synergies between these two data types to improve the quality and depth of the overall proteomic analysis. To this end, we automated the large-scale integration of results from multiprotease bottom-up and top-down analyses in the software program Proteoform Suite and applied it to the analysis of proteoforms from the human Jurkat T lymphocyte cell line. We implemented the recently developed proteoform-level classification scheme for top-down tandem mass spectrometry (MS/MS) identifications in Proteoform Suite, which enables users to observe the level and type of ambiguity for each proteoform identification, including which of the ambiguous proteoform identifications are supported by bottom-up-level evidence. We used Proteoform Suite to find instances where top-down identifications aid in protein inference from bottom-up analysis and conversely where bottom-up peptide identifications aid in proteoform PTM localization. We also show the use of bottom-up data to infer proteoform candidates potentially present in the sample, allowing confirmation of such proteoform candidates by intact-mass analysis of MS1 spectra. The implementation of these capabilities in the freely available software program Proteoform Suite enables users to integrate large-scale top-down and bottom-up data sets and to utilize the synergies between them to improve and extend the proteomic analysis.
中文翻译:

通过自下而上和自上而下数据的集成改进复杂系统中的蛋白质组识别。
细胞功能由大量多样的蛋白质组执行。蛋白质型是由于遗传变异、RNA 剪接和翻译后修饰 (PTM) 而产生的特定蛋白质形式。完整蛋白质的自上而下质谱分析可以识别蛋白质型,包括源自序列切割事件或含有多个 PTM 的蛋白质型。相比之下,自下而上的蛋白质组学鉴定肽,这需要蛋白质推断并且不会产生蛋白质型鉴定。我们在这里寻求利用这两种数据类型之间的协同作用来提高整体蛋白质组学分析的质量和深度。为此,我们在 Proteoform Suite 软件程序中自动对多蛋白酶自下而上和自上而下分析的结果进行了大规模整合,并将其应用于人类 Jurkat T 淋巴细胞细胞系的蛋白质型分析。我们在 Proteoform Suite 中实施了最近开发的用于自上而下串联质谱 (MS/MS) 鉴定的蛋白质型水平分类方案,这使用户能够观察每个蛋白质型鉴定的模糊程度和类型,包括哪些模糊型蛋白质鉴定由自下而上的证据支持。我们使用 Proteoform Suite 来查找自上而下的鉴定有助于从自下而上的分析推断蛋白质的情况,反之,自下而上的肽鉴定有助于蛋白质型 PTM 定位。我们还展示了使用自下而上的数据来推断样品中可能存在的候选蛋白质,从而通过 MS1 光谱的完整质量分析来确认此类候选蛋白质。在免费提供的软件程序 Proteoform Suite 中实施这些功能使用户能够集成大规模自上而下和自下而上的数据集,并利用它们之间的协同作用来改进和扩展蛋白质组学分析。
更新日期:2020-08-08
中文翻译:
通过自下而上和自上而下数据的集成改进复杂系统中的蛋白质组识别。
细胞功能由大量多样的蛋白质组执行。蛋白质型是由于遗传变异、RNA 剪接和翻译后修饰 (PTM) 而产生的特定蛋白质形式。完整蛋白质的自上而下质谱分析可以识别蛋白质型,包括源自序列切割事件或含有多个 PTM 的蛋白质型。相比之下,自下而上的蛋白质组学鉴定肽,这需要蛋白质推断并且不会产生蛋白质型鉴定。我们在这里寻求利用这两种数据类型之间的协同作用来提高整体蛋白质组学分析的质量和深度。为此,我们在 Proteoform Suite 软件程序中自动对多蛋白酶自下而上和自上而下分析的结果进行了大规模整合,并将其应用于人类 Jurkat T 淋巴细胞细胞系的蛋白质型分析。我们在 Proteoform Suite 中实施了最近开发的用于自上而下串联质谱 (MS/MS) 鉴定的蛋白质型水平分类方案,这使用户能够观察每个蛋白质型鉴定的模糊程度和类型,包括哪些模糊型蛋白质鉴定由自下而上的证据支持。我们使用 Proteoform Suite 来查找自上而下的鉴定有助于从自下而上的分析推断蛋白质的情况,反之,自下而上的肽鉴定有助于蛋白质型 PTM 定位。我们还展示了使用自下而上的数据来推断样品中可能存在的候选蛋白质,从而通过 MS1 光谱的完整质量分析来确认此类候选蛋白质。在免费提供的软件程序 Proteoform Suite 中实施这些功能使用户能够集成大规模自上而下和自下而上的数据集,并利用它们之间的协同作用来改进和扩展蛋白质组学分析。










































 京公网安备 11010802027423号
京公网安备 11010802027423号