当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Predicting Hydration Free Energies of the FreeSolv Database of Drug-like Molecules with Molecular Density Functional Theory.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-06-25 , DOI: 10.1021/acs.jcim.0c00526 Sohvi Luukkonen 1 , Luc Belloni 2 , Daniel Borgis 1, 3 , Maximilien Levesque 3, 4
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-06-25 , DOI: 10.1021/acs.jcim.0c00526 Sohvi Luukkonen 1 , Luc Belloni 2 , Daniel Borgis 1, 3 , Maximilien Levesque 3, 4
Affiliation
|
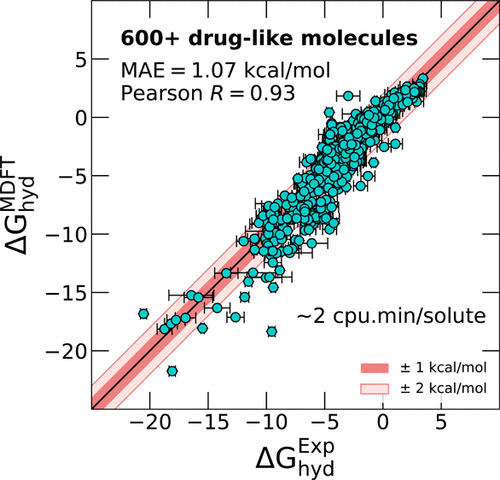
We assess the performance of molecular density functional theory (MDFT) to predict hydration free energies of the small drug-like molecules benchmark, FreeSolv. The MDFT in the hypernetted chain approximation (HNC) coupled with a pressure correction predicts experimental hydration free energies of the FreeSolv database within 1 kcal/mol with an average computation time of 2 cpu·min per molecule. This is the same accuracy as for simulation-based free energy calculations that typically require hundreds of cpu·h or tens of gpu·h per molecule.
中文翻译:

用分子密度泛函理论预测类药物分子的FreeSolv数据库的水合自由能。
我们评估分子密度泛函理论(MDFT)的性能,以预测小类药物分子基准FreeSolv的水合自由能。超网状链逼近(HNC)中的MDFT与压力校正相结合,可预测FreeSolv数据库的实验水合自由能在1 kcal / mol之内,每分子平均计算时间为2 cpu·min。这与基于仿真的自由能计算的精度相同,后者通常每个分子需要数百cpu·h或数十gpu·h。
更新日期:2020-07-27
中文翻译:
用分子密度泛函理论预测类药物分子的FreeSolv数据库的水合自由能。
我们评估分子密度泛函理论(MDFT)的性能,以预测小类药物分子基准FreeSolv的水合自由能。超网状链逼近(HNC)中的MDFT与压力校正相结合,可预测FreeSolv数据库的实验水合自由能在1 kcal / mol之内,每分子平均计算时间为2 cpu·min。这与基于仿真的自由能计算的精度相同,后者通常每个分子需要数百cpu·h或数十gpu·h。










































 京公网安备 11010802027423号
京公网安备 11010802027423号