当前位置:
X-MOL 学术
›
J. Leukoc. Biol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Targeting NLRP3 and staphylococcal pore-forming toxin receptors in human-induced pluripotent stem cell-derived macrophages.
Journal of Leukocyte Biology ( IF 3.6 ) Pub Date : 2020-06-12 , DOI: 10.1002/jlb.4ma0420-497r Seong H Chow 1 , Pankaj Deo 1 , Amy T Y Yeung 2 , Xenia P Kostoulias 3 , Yusun Jeon 1 , Mei-Ling Gao 4, 5 , Azadeh Seidi 1 , Françios Alwyn Benson Olivier 1 , Sushmita Sridhar 2 , Cara Nethercott 3 , David Cameron 3 , Avril A B Robertson 6 , Remy Robert 1 , Charles R Mackay 3 , Ana Traven 1 , Zi-Bing Jin 4, 5 , Christine Hale 2 , Gordon Dougan 2, 7 , Anton Y Peleg 3, 8 , Thomas Naderer 1
Journal of Leukocyte Biology ( IF 3.6 ) Pub Date : 2020-06-12 , DOI: 10.1002/jlb.4ma0420-497r Seong H Chow 1 , Pankaj Deo 1 , Amy T Y Yeung 2 , Xenia P Kostoulias 3 , Yusun Jeon 1 , Mei-Ling Gao 4, 5 , Azadeh Seidi 1 , Françios Alwyn Benson Olivier 1 , Sushmita Sridhar 2 , Cara Nethercott 3 , David Cameron 3 , Avril A B Robertson 6 , Remy Robert 1 , Charles R Mackay 3 , Ana Traven 1 , Zi-Bing Jin 4, 5 , Christine Hale 2 , Gordon Dougan 2, 7 , Anton Y Peleg 3, 8 , Thomas Naderer 1
Affiliation
|
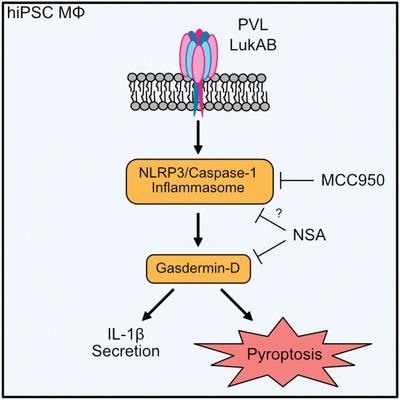
Staphylococcus aureus causes necrotizing pneumonia by secreting toxins such as leukocidins that target front‐line immune cells. The mechanism by which leukocidins kill innate immune cells and trigger inflammation during S. aureus lung infection, however, remains unresolved. Here, we explored human‐induced pluripotent stem cell‐derived macrophages (hiPSC‐dMs) to study the interaction of the leukocidins Panton–Valentine leukocidin (PVL) and LukAB with lung macrophages, which are the initial leukocidin targets during S. aureus lung invasion. hiPSC‐dMs were susceptible to the leukocidins PVL and LukAB and both leukocidins triggered NLPR3 inflammasome activation resulting in IL‐1β secretion. hiPSC‐dM cell death after LukAB exposure, however, was only temporarily dependent of NLRP3, although NLRP3 triggered marked cell death after PVL treatment. CRISPR/Cas9‐mediated deletion of the PVL receptor, C5aR1, protected hiPSC‐dMs from PVL cytotoxicity, despite the expression of other leukocidin receptors, such as CD45. PVL‐deficient S. aureus had reduced ability to induce lung IL‐1β levels in human C5aR1 knock‐in mice. Unexpectedly, inhibiting NLRP3 activity resulted in increased wild‐type S. aureus lung burdens. Our findings suggest that NLRP3 induces macrophage death and IL‐1β secretion after PVL exposure and controls S. aureus lung burdens.
中文翻译:

在人类诱导的多能干细胞衍生的巨噬细胞中靶向NLRP3和葡萄球菌成孔毒素受体。
金黄色葡萄球菌通过分泌针对前线免疫细胞的毒素(如白细胞介素)而导致坏死性肺炎。然而,白血球菌素杀死金黄色葡萄球菌肺部感染期间先天免疫细胞并引发炎症的机制仍未阐明。在这里,我们探索了人类诱导的多能干细胞衍生的巨噬细胞(hiPSC-dMs),以研究白细胞结合素Panton-Valentine白细胞结合素(PVL)和LukAB与肺巨噬细胞的相互作用,这是金黄色葡萄球菌最初的白细胞结合素靶点。肺部侵袭。hiPSC-dM易受白介素PVL和LukAB的影响,两种白介素均触发NLPR3炎性体激活,导致IL-1β分泌。尽管在PVL处理后NLRP3触发了明显的细胞死亡,但LukAB暴露后的hiPSC-dM细胞死亡只是暂时依赖于NLRP3。CRISPR / Cas9介导的PVL受体C5aR1的缺失使hiPSC-dMs免受PVL细胞毒性,尽管其他白细胞抑制素受体(如CD45)也表达了这种表达。缺乏PVL的金黄色葡萄球菌在人类C5aR1敲入小鼠中诱导肺IL-1β水平降低的能力。出乎意料的是,抑制NLRP3活性导致野生型金黄色葡萄球菌增加肺部负担。我们的发现表明,NLRP3诱导PVL暴露后巨噬细胞死亡和IL-1β分泌,并控制金黄色葡萄球菌的肺负担。
更新日期:2020-06-12
中文翻译:
在人类诱导的多能干细胞衍生的巨噬细胞中靶向NLRP3和葡萄球菌成孔毒素受体。
金黄色葡萄球菌通过分泌针对前线免疫细胞的毒素(如白细胞介素)而导致坏死性肺炎。然而,白血球菌素杀死金黄色葡萄球菌肺部感染期间先天免疫细胞并引发炎症的机制仍未阐明。在这里,我们探索了人类诱导的多能干细胞衍生的巨噬细胞(hiPSC-dMs),以研究白细胞结合素Panton-Valentine白细胞结合素(PVL)和LukAB与肺巨噬细胞的相互作用,这是金黄色葡萄球菌最初的白细胞结合素靶点。肺部侵袭。hiPSC-dM易受白介素PVL和LukAB的影响,两种白介素均触发NLPR3炎性体激活,导致IL-1β分泌。尽管在PVL处理后NLRP3触发了明显的细胞死亡,但LukAB暴露后的hiPSC-dM细胞死亡只是暂时依赖于NLRP3。CRISPR / Cas9介导的PVL受体C5aR1的缺失使hiPSC-dMs免受PVL细胞毒性,尽管其他白细胞抑制素受体(如CD45)也表达了这种表达。缺乏PVL的金黄色葡萄球菌在人类C5aR1敲入小鼠中诱导肺IL-1β水平降低的能力。出乎意料的是,抑制NLRP3活性导致野生型金黄色葡萄球菌增加肺部负担。我们的发现表明,NLRP3诱导PVL暴露后巨噬细胞死亡和IL-1β分泌,并控制金黄色葡萄球菌的肺负担。








































 京公网安备 11010802027423号
京公网安备 11010802027423号