当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The Discovery of a Putative Allosteric Site in the SARS-CoV-2 Spike Protein Using an Integrated Structural/Dynamic Approach.
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2020-06-17 , DOI: 10.1021/acs.jproteome.0c00273 Luisa Di Paola 1 , Hamid Hadi-Alijanvand 2 , Xingyu Song 3 , Guang Hu 3 , Alessandro Giuliani 4
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2020-06-17 , DOI: 10.1021/acs.jproteome.0c00273 Luisa Di Paola 1 , Hamid Hadi-Alijanvand 2 , Xingyu Song 3 , Guang Hu 3 , Alessandro Giuliani 4
Affiliation
|
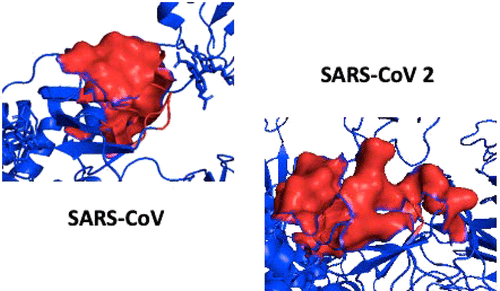
SARS-CoV-2 has caused the largest pandemic of the twenty-first century (COVID-19), threatening the life and economy of all countries in the world. The identification of novel therapies and vaccines that can mitigate or control this global health threat is among the most important challenges facing biomedical sciences. To construct a long-term strategy to fight both SARS-CoV-2 and other possible future threats from coronaviruses, it is critical to understand the molecular mechanisms underlying the virus action. The viral entry and associated infectivity stems from the formation of the SARS-CoV-2 spike protein complex with angiotensin-converting enzyme 2 (ACE2). The detection of putative allosteric sites on the viral spike protein molecule can be used to elucidate the molecular pathways that can be targeted with allosteric drugs to weaken the spike-ACE2 interaction and, thus, reduce viral infectivity. In this study, we present the results of the application of different computational methods aimed at detecting allosteric sites on the SARS-CoV-2 spike protein. The adopted tools consisted of the protein contact networks (PCNs), SEPAS (Affinity by Flexibility), and perturbation response scanning (PRS) based on elastic network modes. All of these methods were applied to the ACE2 complex with both the SARS-CoV2 and SARS-CoV spike proteins. All of the adopted analyses converged toward a specific region (allosteric modulation region [AMR]), present in both complexes and predicted to act as an allosteric site modulating the binding of the spike protein with ACE2. Preliminary results on hepcidin (a molecule with strong structural and sequence with AMR) indicated an inhibitory effect on the binding affinity of the spike protein toward the ACE2 protein.
中文翻译:

使用综合结构/动态方法发现 SARS-CoV-2 刺突蛋白中假定的变构位点。
SARS-CoV-2引发了二十一世纪最大规模的大流行(COVID-19),威胁着世界各国的生命和经济。确定能够减轻或控制这一全球健康威胁的新疗法和疫苗是生物医学科学面临的最重要挑战之一。为了制定对抗 SARS-CoV-2 和冠状病毒未来可能带来的其他威胁的长期战略,了解病毒作用背后的分子机制至关重要。病毒的进入和相关的感染性源于 SARS-CoV-2 刺突蛋白复合物与血管紧张素转换酶 2 (ACE2) 的形成。病毒刺突蛋白分子上假定的变构位点的检测可用于阐明变构药物可靶向的分子途径,以削弱刺突-ACE2相互作用,从而降低病毒感染性。在这项研究中,我们展示了应用不同计算方法的结果,旨在检测 SARS-CoV-2 刺突蛋白上的变构位点。采用的工具包括蛋白质接触网络(PCN)、SEPAS(Affinity by Flexibility)和基于弹性网络模式的微扰响应扫描(PRS)。所有这些方法均应用于 ACE2 与 SARS-CoV2 和 SARS-CoV 刺突蛋白的复合物。所有采用的分析都集中于一个特定区域(变构调节区域 [AMR]),该区域存在于两种复合物中,并预计将作为调节刺突蛋白与 ACE2 结合的变构位点。铁调素(一种具有强结构和 AMR 序列的分子)的初步结果表明,它对刺突蛋白与 ACE2 蛋白的结合亲和力有抑制作用。
更新日期:2020-06-17
中文翻译:
使用综合结构/动态方法发现 SARS-CoV-2 刺突蛋白中假定的变构位点。
SARS-CoV-2引发了二十一世纪最大规模的大流行(COVID-19),威胁着世界各国的生命和经济。确定能够减轻或控制这一全球健康威胁的新疗法和疫苗是生物医学科学面临的最重要挑战之一。为了制定对抗 SARS-CoV-2 和冠状病毒未来可能带来的其他威胁的长期战略,了解病毒作用背后的分子机制至关重要。病毒的进入和相关的感染性源于 SARS-CoV-2 刺突蛋白复合物与血管紧张素转换酶 2 (ACE2) 的形成。病毒刺突蛋白分子上假定的变构位点的检测可用于阐明变构药物可靶向的分子途径,以削弱刺突-ACE2相互作用,从而降低病毒感染性。在这项研究中,我们展示了应用不同计算方法的结果,旨在检测 SARS-CoV-2 刺突蛋白上的变构位点。采用的工具包括蛋白质接触网络(PCN)、SEPAS(Affinity by Flexibility)和基于弹性网络模式的微扰响应扫描(PRS)。所有这些方法均应用于 ACE2 与 SARS-CoV2 和 SARS-CoV 刺突蛋白的复合物。所有采用的分析都集中于一个特定区域(变构调节区域 [AMR]),该区域存在于两种复合物中,并预计将作为调节刺突蛋白与 ACE2 结合的变构位点。铁调素(一种具有强结构和 AMR 序列的分子)的初步结果表明,它对刺突蛋白与 ACE2 蛋白的结合亲和力有抑制作用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号