当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Interface kinetics of rapid solidification of binary alloys by atomistic simulations: Application to Ti-Ni alloys
Computational Materials Science ( IF 3.1 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.commatsci.2020.109854 Sepideh Kavousi , Brian R. Novak , Jeffrey Hoyt , Dorel Moldovan
Computational Materials Science ( IF 3.1 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.commatsci.2020.109854 Sepideh Kavousi , Brian R. Novak , Jeffrey Hoyt , Dorel Moldovan
|
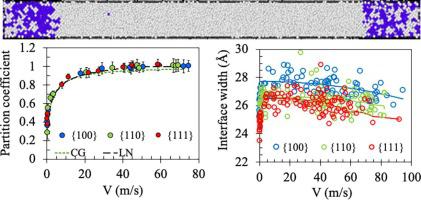
Abstract Using molecular dynamics and Monte Carlo simulations we investigate the non-equilibrium interfacial kinetics during rapid solidification of Ti-Ni alloys. According to the existing theories, the kinetic coefficient is related, via an analytical expression, to the equilibrium and non-equilibrium solute concentration profiles across the crystal-melt interface, the interface temperature and velocity, and the drag coefficient. The kinetic coefficient was obtained by deriving these properties from specifically designed atomistic simulations and then fitting using the analytical expression. The results show that the kinetic coefficient is only weakly anisotropic and increases with increasing temperature. The velocity-dependent partition coefficient, as described by two solute trapping models, the continuous growth and the local non-equilibrium models, were fitted to the molecular dynamics simulation results. In addition, molecular dynamics and semi-grand canonical Monte Carlo simulations suggest that complete solute trapping might occur only under certain conditions. This can be explained by the fact that the maximum achievable effective free energy driving force for solidification, which defines the complete solute trapping, is limited by the chemical potential and free energy profiles for the Ti-Ni alloy. The investigations, using molecular dynamics simulations, of the dependence of crystal-melt interface width on solidification velocity show, at high velocities, similar trend to that predicted by the hyperbolic phase field model which is suitable for studies of rapid solidification of alloys.
中文翻译:

基于原子模拟的二元合金快速凝固界面动力学:在 Ti-Ni 合金中的应用
摘要 利用分子动力学和蒙特卡罗模拟,我们研究了 Ti-Ni 合金快速凝固过程中的非平衡界面动力学。根据现有理论,动力学系数通过解析表达式与晶体-熔体界面上的平衡和非平衡溶质浓度分布、界面温度和速度以及阻力系数相关。动力学系数是通过从专门设计的原子模拟中推导出这些特性,然后使用解析表达式进行拟合而获得的。结果表明,动力学系数只是微弱的各向异性,并且随着温度的升高而增加。速度相关的分配系数,如两个溶质捕获模型所描述的,连续增长和局部非平衡模型与分子动力学模拟结果相吻合。此外,分子动力学和半正则蒙特卡罗模拟表明,完全的溶质捕获可能仅在某些条件下发生。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。符合分子动力学模拟结果。此外,分子动力学和半正则蒙特卡罗模拟表明,完全的溶质捕获可能仅在某些条件下发生。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。符合分子动力学模拟结果。此外,分子动力学和半正则蒙特卡罗模拟表明,完全的溶质捕获可能仅在某些条件下发生。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。分子动力学和半正则蒙特卡罗模拟表明,完全的溶质捕获可能仅在某些条件下发生。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。分子动力学和半正则蒙特卡罗模拟表明,完全的溶质捕获可能仅在某些条件下发生。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。
更新日期:2020-11-01
中文翻译:
基于原子模拟的二元合金快速凝固界面动力学:在 Ti-Ni 合金中的应用
摘要 利用分子动力学和蒙特卡罗模拟,我们研究了 Ti-Ni 合金快速凝固过程中的非平衡界面动力学。根据现有理论,动力学系数通过解析表达式与晶体-熔体界面上的平衡和非平衡溶质浓度分布、界面温度和速度以及阻力系数相关。动力学系数是通过从专门设计的原子模拟中推导出这些特性,然后使用解析表达式进行拟合而获得的。结果表明,动力学系数只是微弱的各向异性,并且随着温度的升高而增加。速度相关的分配系数,如两个溶质捕获模型所描述的,连续增长和局部非平衡模型与分子动力学模拟结果相吻合。此外,分子动力学和半正则蒙特卡罗模拟表明,完全的溶质捕获可能仅在某些条件下发生。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。符合分子动力学模拟结果。此外,分子动力学和半正则蒙特卡罗模拟表明,完全的溶质捕获可能仅在某些条件下发生。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。符合分子动力学模拟结果。此外,分子动力学和半正则蒙特卡罗模拟表明,完全的溶质捕获可能仅在某些条件下发生。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。分子动力学和半正则蒙特卡罗模拟表明,完全的溶质捕获可能仅在某些条件下发生。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。分子动力学和半正则蒙特卡罗模拟表明,完全的溶质捕获可能仅在某些条件下发生。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。这可以通过以下事实来解释:凝固的最大可实现有效自由能驱动力(定义完全溶质俘获)受 Ti-Ni 合金的化学势和自由能分布的限制。使用分子动力学模拟对晶体-熔体界面宽度对凝固速度的依赖性的研究表明,在高速下,与适用于合金快速凝固研究的双曲线相场模型预测的趋势相似。










































 京公网安备 11010802027423号
京公网安备 11010802027423号