当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Accurate Prediction of GPCR Ligand Binding Affinity with Free Energy Perturbation.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-06-15 , DOI: 10.1021/acs.jcim.0c00449 Francesca Deflorian 1 , Laura Perez-Benito 2 , Eelke B Lenselink 3 , Miles Congreve 1 , Herman W T van Vlijmen 2 , Jonathan S Mason 1 , Chris de Graaf 1 , Gary Tresadern 2
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-06-15 , DOI: 10.1021/acs.jcim.0c00449 Francesca Deflorian 1 , Laura Perez-Benito 2 , Eelke B Lenselink 3 , Miles Congreve 1 , Herman W T van Vlijmen 2 , Jonathan S Mason 1 , Chris de Graaf 1 , Gary Tresadern 2
Affiliation
|
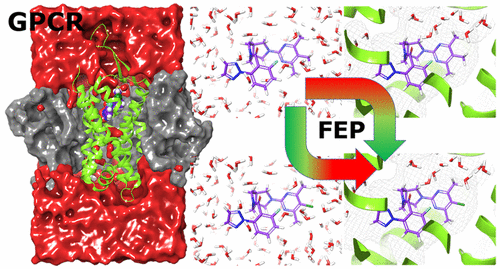
The computational prediction of relative binding free energies is a crucial goal for drug discovery, and G protein-coupled receptors (GPCRs) are arguably the most important drug target class. However, they present increased complexity to model compared to soluble globular proteins. Despite breakthroughs, experimental X-ray crystal and cryo-EM structures are challenging to attain, meaning computational models of the receptor and ligand binding mode are sometimes necessary. This leads to uncertainty in understanding ligand–protein binding induced changes such as, water positioning and displacement, side chain positioning, hydrogen bond networks, and the overall structure of the hydration shell around the ligand and protein. In other words, the very elements that define structure activity relationships (SARs) and are crucial for accurate binding free energy calculations are typically more uncertain for GPCRs. In this work we use free energy perturbation (FEP) to predict the relative binding free energies for ligands of two different GPCRs. We pinpoint the key aspects for success such as the important role of key water molecules, amino acid ionization states, and the benefit of equilibration with specific ligands. Initial calculations following typical FEP setup and execution protocols delivered no correlation with experiment, but we show how results are improved in a logical and systematic way. This approach gave, in the best cases, a coefficient of determination (R2) compared with experiment in the range of 0.6–0.9 and mean unsigned errors compared to experiment of 0.6–0.7 kcal/mol. We anticipate that our findings will be applicable to other difficult-to-model protein ligand data sets and be of wide interest for the community to continue improving FE binding energy predictions.
中文翻译:

准确预测GPCR配体结合亲和力与自由能干扰。
相对结合自由能的计算预测是药物发现的关键目标,而G蛋白偶联受体(GPCR)无疑是最重要的药物靶标类别。但是,与可溶性球状蛋白相比,它们呈现出更高的建模复杂性。尽管取得了突破,但实验性X射线晶体和cryo-EM结构仍难以实现,这意味着有时需要受体和配体结合模式的计算模型。这导致无法理解配体与蛋白质结合引起的变化,例如水的位置和位移,侧链定位,氢键网络以及配体和蛋白质周围水合壳的整体结构。换一种说法,对于GPCR而言,定义结构活性关系(SAR)且对于精确结合自由能计算至关重要的元素通常更加不确定。在这项工作中,我们使用自由能扰动(FEP)来预测两个不同GPCR配体的相对结合自由能。我们指出了成功的关键方面,例如关键水分子的重要作用,氨基酸电离状态以及与特定配体平衡的好处。遵循典型FEP设置和执行协议的初始计算与实验没有相关性,但是我们展示了如何以逻辑和系统的方式改善结果。在最佳情况下,此方法可得出确定系数(在这项工作中,我们使用自由能扰动(FEP)来预测两个不同GPCR配体的相对结合自由能。我们指出了成功的关键方面,例如关键水分子的重要作用,氨基酸电离状态以及与特定配体平衡的好处。遵循典型FEP设置和执行协议的初始计算与实验没有相关性,但是我们展示了如何以逻辑和系统的方式改善结果。在最佳情况下,此方法可得出确定系数(在这项工作中,我们使用自由能扰动(FEP)来预测两个不同GPCR配体的相对结合自由能。我们指出了成功的关键方面,例如关键水分子的重要作用,氨基酸电离状态以及与特定配体平衡的好处。遵循典型FEP设置和执行协议的初始计算与实验没有相关性,但是我们展示了如何以逻辑和系统的方式改善结果。在最佳情况下,此方法可得出确定系数(遵循典型FEP设置和执行协议的初始计算与实验没有相关性,但是我们展示了如何以逻辑和系统的方式改善结果。在最佳情况下,此方法可得出确定系数(遵循典型FEP设置和执行协议的初始计算与实验没有相关性,但是我们展示了如何以逻辑和系统的方式改善结果。在最佳情况下,此方法可得出确定系数(R 2)与实验范围在0.6–0.9之间,平均无符号误差与实验范围在0.6–0.7 kcal / mol之间。我们预计我们的发现将适用于其他难以建模的蛋白质配体数据集,并且对于社区继续改善FE结合能的预测具有广泛的兴趣。
更新日期:2020-06-15
中文翻译:
准确预测GPCR配体结合亲和力与自由能干扰。
相对结合自由能的计算预测是药物发现的关键目标,而G蛋白偶联受体(GPCR)无疑是最重要的药物靶标类别。但是,与可溶性球状蛋白相比,它们呈现出更高的建模复杂性。尽管取得了突破,但实验性X射线晶体和cryo-EM结构仍难以实现,这意味着有时需要受体和配体结合模式的计算模型。这导致无法理解配体与蛋白质结合引起的变化,例如水的位置和位移,侧链定位,氢键网络以及配体和蛋白质周围水合壳的整体结构。换一种说法,对于GPCR而言,定义结构活性关系(SAR)且对于精确结合自由能计算至关重要的元素通常更加不确定。在这项工作中,我们使用自由能扰动(FEP)来预测两个不同GPCR配体的相对结合自由能。我们指出了成功的关键方面,例如关键水分子的重要作用,氨基酸电离状态以及与特定配体平衡的好处。遵循典型FEP设置和执行协议的初始计算与实验没有相关性,但是我们展示了如何以逻辑和系统的方式改善结果。在最佳情况下,此方法可得出确定系数(在这项工作中,我们使用自由能扰动(FEP)来预测两个不同GPCR配体的相对结合自由能。我们指出了成功的关键方面,例如关键水分子的重要作用,氨基酸电离状态以及与特定配体平衡的好处。遵循典型FEP设置和执行协议的初始计算与实验没有相关性,但是我们展示了如何以逻辑和系统的方式改善结果。在最佳情况下,此方法可得出确定系数(在这项工作中,我们使用自由能扰动(FEP)来预测两个不同GPCR配体的相对结合自由能。我们指出了成功的关键方面,例如关键水分子的重要作用,氨基酸电离状态以及与特定配体平衡的好处。遵循典型FEP设置和执行协议的初始计算与实验没有相关性,但是我们展示了如何以逻辑和系统的方式改善结果。在最佳情况下,此方法可得出确定系数(遵循典型FEP设置和执行协议的初始计算与实验没有相关性,但是我们展示了如何以逻辑和系统的方式改善结果。在最佳情况下,此方法可得出确定系数(遵循典型FEP设置和执行协议的初始计算与实验没有相关性,但是我们展示了如何以逻辑和系统的方式改善结果。在最佳情况下,此方法可得出确定系数(R 2)与实验范围在0.6–0.9之间,平均无符号误差与实验范围在0.6–0.7 kcal / mol之间。我们预计我们的发现将适用于其他难以建模的蛋白质配体数据集,并且对于社区继续改善FE结合能的预测具有广泛的兴趣。










































 京公网安备 11010802027423号
京公网安备 11010802027423号