Chem ( IF 19.1 ) Pub Date : 2020-06-16 , DOI: 10.1016/j.chempr.2020.05.014 Mojtaba Haghighatlari 1 , Jie Li 1 , Farnaz Heidar-Zadeh 1, 2, 3 , Yuchen Liu 1 , Xingyi Guan 1 , Teresa Head-Gordon 1, 4, 5
|
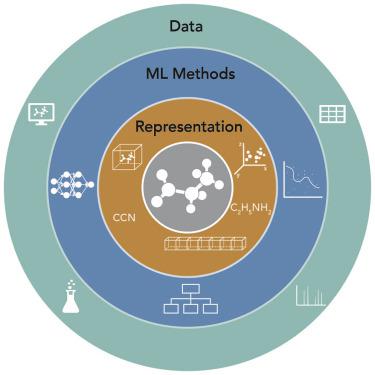
Recently, supervised machine learning has been ascending in providing new predictive approaches for chemical, biological, and materials sciences applications. In this Perspective, we focus on the interplay of machine learning methods with the chemically motivated descriptors and the size and type of datasets needed for molecular property prediction. Using nuclear magnetic resonance chemical shift prediction as an example, we demonstrate that success is predicated on the choice of feature extracted or real-space representations of chemical structures, whether the molecular property data are abundant and/or experimentally or computationally derived, and how these together will influence the correct choice of popular machine learning methods drawn from deep learning, random forests, or kernel methods.
中文翻译:
学习进行化学预测:特征表示、数据和机器学习方法的相互作用。
最近,监督机器学习在为化学、生物和材料科学应用提供新的预测方法方面不断进步。在这个视角中,我们重点关注机器学习方法与化学驱动描述符以及分子特性预测所需的数据集的大小和类型的相互作用。以核磁共振化学位移预测为例,我们证明成功取决于化学结构的特征提取或真实空间表示的选择,分子特性数据是否丰富和/或通过实验或计算得出,以及这些数据如何获得共同将影响对深度学习、随机森林或核方法中流行的机器学习方法的正确选择。










































 京公网安备 11010802027423号
京公网安备 11010802027423号