当前位置:
X-MOL 学术
›
Int. J. Quantum Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A density functional theory study on the atmospheric reaction of CH3O2 with HS: Mechanism and kinetics
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2020-06-13 , DOI: 10.1002/qua.26330 Shiguo Zhang 1 , Yan Zhang 1 , Yun Zhang 1 , Ziyan Feng 1 , Caihong Wang 1 , He Bian 1 , Jinshe Chen 2
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2020-06-13 , DOI: 10.1002/qua.26330 Shiguo Zhang 1 , Yan Zhang 1 , Yun Zhang 1 , Ziyan Feng 1 , Caihong Wang 1 , He Bian 1 , Jinshe Chen 2
Affiliation
|
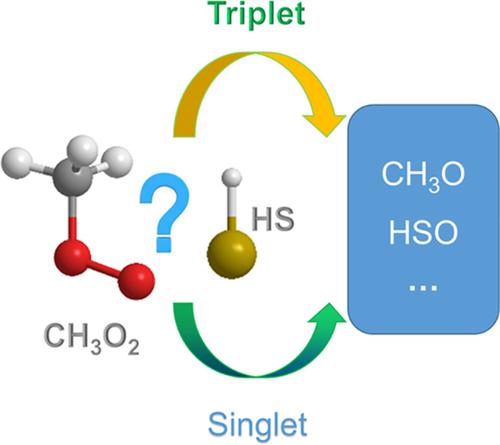
The reaction mechanism of CH3O2 and HS was systematically investigated by density functional theory (DFT). Six singlet pathways and seven triplet ones are located on the potential surface (PES). The result indicates that the main products are CH3O and HSO both on the singlet and triplet PES, different from the CH3O2 + OH reaction. Moreover, deformation density (ρ def) and atoms in molecules (AIM) analyses were carried out to further uncover the nature of chemical bonding evolution in the primary pathways. Furthermore, reaction rate constants were calculated in the temperature range from 200 to 1000 K using the transition state theory with the Wigner and Eckart tunneling corrections. Our results can shed light on the title reaction and offer instructions for analogous atmospheric reactions, as well as experimental research in the future.
中文翻译:

CH3O2与HS的大气反应的密度泛函理论研究:机理和动力学
利用密度泛函理论(DFT)系统地研究了CH 3 O 2与HS的反应机理。六个单线态路径和七个三线态路径位于潜在表面(PES)上。结果表明,与CH 3 O 2 + OH反应不同,单峰和三峰PES上的主要产物均为CH 3 O和HSO 。此外,变形密度(ρ DEF)和分子中的原子(AIM)分析,以进一步揭示主要途径中化学键演化的性质。此外,使用过渡态理论和Wigner和Eckart隧穿校正,在200至1000 K的温度范围内计算了反应速率常数。我们的结果可以阐明标题反应,并提供类似大气反应的指示以及将来的实验研究。
更新日期:2020-07-20
中文翻译:
CH3O2与HS的大气反应的密度泛函理论研究:机理和动力学
利用密度泛函理论(DFT)系统地研究了CH 3 O 2与HS的反应机理。六个单线态路径和七个三线态路径位于潜在表面(PES)上。结果表明,与CH 3 O 2 + OH反应不同,单峰和三峰PES上的主要产物均为CH 3 O和HSO 。此外,变形密度(ρ DEF)和分子中的原子(AIM)分析,以进一步揭示主要途径中化学键演化的性质。此外,使用过渡态理论和Wigner和Eckart隧穿校正,在200至1000 K的温度范围内计算了反应速率常数。我们的结果可以阐明标题反应,并提供类似大气反应的指示以及将来的实验研究。










































 京公网安备 11010802027423号
京公网安备 11010802027423号