当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational Determination of Potential Inhibitors of SARS-CoV-2 Main Protease.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-06-12 , DOI: 10.1021/acs.jcim.0c00491 Son Tung Ngo 1, 2 , Ngoc Quynh Anh Pham 3 , Ly Thi Le 4 , Duc-Hung Pham 5 , Van V Vu 6
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-06-12 , DOI: 10.1021/acs.jcim.0c00491 Son Tung Ngo 1, 2 , Ngoc Quynh Anh Pham 3 , Ly Thi Le 4 , Duc-Hung Pham 5 , Van V Vu 6
Affiliation
|
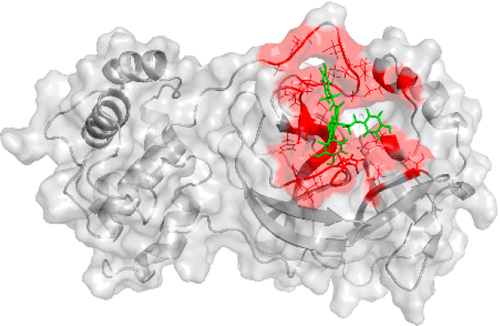
The novel coronavirus (SARS-CoV-2) has infected several million people and caused thousands of deaths worldwide since December 2019. As the disease is spreading rapidly all over the world, it is urgent to find effective drugs to treat the virus. The main protease (Mpro) of SARS-CoV-2 is one of the potential drug targets. Therefore, in this context, we used rigorous computational methods, including molecular docking, fast pulling of ligand (FPL), and free energy perturbation (FEP), to investigate potential inhibitors of SARS-CoV-2 Mpro. We first tested our approach with three reported inhibitors of SARS-CoV-2 Mpro, and our computational results are in good agreement with the respective experimental data. Subsequently, we applied our approach on a database of ∼4600 natural compounds, as well as 8 available HIV-1 protease (PR) inhibitors and an aza-peptide epoxide. Molecular docking resulted in a short list of 35 natural compounds, which was subsequently refined using the FPL scheme. FPL simulations resulted in five potential inhibitors, including three natural compounds and two available HIV-1 PR inhibitors. Finally, FEP, the most accurate and precise method, was used to determine the absolute binding free energy of these five compounds. FEP results indicate that two natural compounds, cannabisin A and isoacteoside, and an HIV-1 PR inhibitor, darunavir, exhibit a large binding free energy to SARS-CoV-2 Mpro, which is larger than that of 13b, the most reliable SARS-CoV-2 Mpro inhibitor recently reported. The binding free energy largely arises from van der Waals interaction. We also found that Glu166 forms H-bonds to all of the inhibitors. Replacing Glu166 by an alanine residue leads to ∼2.0 kcal/mol decreases in the affinity of darunavir to SARS-CoV-2 Mpro. Our results could contribute to the development of potential drugs inhibiting SARS-CoV-2.
中文翻译:

SARS-CoV-2 主要蛋白酶的潜在抑制剂的计算确定。
自2019年12月以来,新型冠状病毒(SARS-CoV-2)已在全球范围内感染数百万人并导致数千人死亡。随着该疾病在世界各地迅速传播,迫切需要找到有效的药物来治疗该病毒。SARS-CoV-2的主要蛋白酶(Mpro)是潜在的药物靶点之一。因此,在此背景下,我们使用严格的计算方法,包括分子对接、配体快速拉动(FPL)和自由能扰动(FEP),来研究 SARS-CoV-2 Mpro 的潜在抑制剂。我们首先用三种报道的 SARS-CoV-2 Mpro 抑制剂测试了我们的方法,我们的计算结果与各自的实验数据非常吻合。随后,我们将我们的方法应用于约 4600 种天然化合物以及 8 种可用的 HIV-1 蛋白酶 (PR) 抑制剂和氮杂肽环氧化物的数据库。分子对接产生了 35 种天然化合物的简短清单,随后使用 FPL 方案进行了完善。FPL 模拟产生了五种潜在的抑制剂,包括三种天然化合物和两种可用的 HIV-1 PR 抑制剂。最后,使用最准确、最精密的方法 FEP 测定了这五种化合物的绝对结合自由能。FEP 结果表明,两种天然化合物大麻素 A 和异类叶升麻苷以及 HIV-1 PR 抑制剂达芦那韦对 SARS-CoV-2 Mpro 表现出很大的结合自由能,比最可靠的 SARS -CoV- 13b的结合自由能更大。最近报道了 CoV-2 Mpro 抑制剂。结合自由能主要来自范德华相互作用。我们还发现 Glu166 与所有抑制剂形成氢键。用丙氨酸残基替换 Glu166 会导致达芦那韦与 SARS-CoV-2 Mpro 的亲和力降低约 2.0 kcal/mol。我们的结果可能有助于开发抑制 SARS-CoV-2 的潜在药物。
更新日期:2020-06-12
中文翻译:
SARS-CoV-2 主要蛋白酶的潜在抑制剂的计算确定。
自2019年12月以来,新型冠状病毒(SARS-CoV-2)已在全球范围内感染数百万人并导致数千人死亡。随着该疾病在世界各地迅速传播,迫切需要找到有效的药物来治疗该病毒。SARS-CoV-2的主要蛋白酶(Mpro)是潜在的药物靶点之一。因此,在此背景下,我们使用严格的计算方法,包括分子对接、配体快速拉动(FPL)和自由能扰动(FEP),来研究 SARS-CoV-2 Mpro 的潜在抑制剂。我们首先用三种报道的 SARS-CoV-2 Mpro 抑制剂测试了我们的方法,我们的计算结果与各自的实验数据非常吻合。随后,我们将我们的方法应用于约 4600 种天然化合物以及 8 种可用的 HIV-1 蛋白酶 (PR) 抑制剂和氮杂肽环氧化物的数据库。分子对接产生了 35 种天然化合物的简短清单,随后使用 FPL 方案进行了完善。FPL 模拟产生了五种潜在的抑制剂,包括三种天然化合物和两种可用的 HIV-1 PR 抑制剂。最后,使用最准确、最精密的方法 FEP 测定了这五种化合物的绝对结合自由能。FEP 结果表明,两种天然化合物大麻素 A 和异类叶升麻苷以及 HIV-1 PR 抑制剂达芦那韦对 SARS-CoV-2 Mpro 表现出很大的结合自由能,比最可靠的 SARS -CoV- 13b的结合自由能更大。最近报道了 CoV-2 Mpro 抑制剂。结合自由能主要来自范德华相互作用。我们还发现 Glu166 与所有抑制剂形成氢键。用丙氨酸残基替换 Glu166 会导致达芦那韦与 SARS-CoV-2 Mpro 的亲和力降低约 2.0 kcal/mol。我们的结果可能有助于开发抑制 SARS-CoV-2 的潜在药物。










































 京公网安备 11010802027423号
京公网安备 11010802027423号