当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Capturing Protein-Ligand Recognition Pathways in Coarse-Grained Simulation.
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2020-06-10 , DOI: 10.1021/acs.jpclett.0c01683 Bhupendra R Dandekar 1 , Jagannath Mondal 1
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2020-06-10 , DOI: 10.1021/acs.jpclett.0c01683 Bhupendra R Dandekar 1 , Jagannath Mondal 1
Affiliation
|
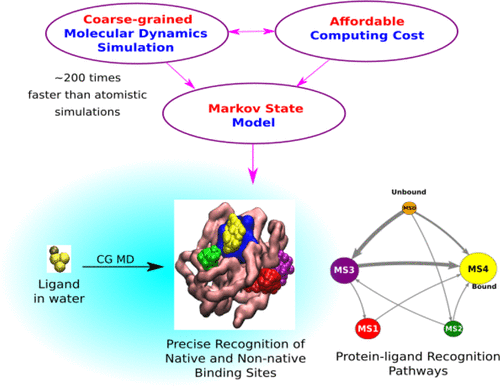
Protein–ligand recognition is dynamic and complex. A key approach in deciphering the mechanism underlying the recognition process is to capture the kinetic process of the ligand in its act of binding to its designated protein cavity. Toward this end, ultralong all-atom molecular dynamics simulation has recently emerged as a popular method of choice because of its ability to record these events at high spatial and temporal resolution. However, success via this route comes at an exorbitant computational cost. Herein, we demonstrate that coarse-grained models of the protein, when systematically optimized to maintain its tertiary fold, can capture the complete process of spontaneous protein–ligand binding from bulk media to the cavity at crystallographic precision and within wall clock time that is orders of magnitude shorter than that of all-atom simulations. The exhaustive sampling of ligand exploration in protein and solvent, harnessed by coarse-grained simulation, leads to elucidation of new ligand recognition pathways and discovery of non-native binding poses.
中文翻译:

在粗粒度模拟中捕获蛋白质-配体识别途径。
蛋白质-配体识别是动态且复杂的。破译识别过程基础的关键方法是捕获配体与其指定蛋白腔结合的动力学过程。为此,超长全原子分子动力学模拟最近已成为一种流行的选择方法,因为它能够以高时空分辨率记录这些事件。但是,通过这种途径获得成功需要付出高昂的计算成本。在此,我们证明了该蛋白质的粗粒度模型,经过系统优化以维持其三级折叠时,可以捕获完整的自发性蛋白质-配体从晶体到腔体的完整结合过程,且具有晶体学精确度,且在壁钟时间内比全原子模拟要短几个数量级。通过粗粒度模拟,对蛋白质和溶剂中的配体探索进行了详尽的采样,从而阐明了新的配体识别途径并发现了非天然结合姿势。
更新日期:2020-07-02
中文翻译:
在粗粒度模拟中捕获蛋白质-配体识别途径。
蛋白质-配体识别是动态且复杂的。破译识别过程基础的关键方法是捕获配体与其指定蛋白腔结合的动力学过程。为此,超长全原子分子动力学模拟最近已成为一种流行的选择方法,因为它能够以高时空分辨率记录这些事件。但是,通过这种途径获得成功需要付出高昂的计算成本。在此,我们证明了该蛋白质的粗粒度模型,经过系统优化以维持其三级折叠时,可以捕获完整的自发性蛋白质-配体从晶体到腔体的完整结合过程,且具有晶体学精确度,且在壁钟时间内比全原子模拟要短几个数量级。通过粗粒度模拟,对蛋白质和溶剂中的配体探索进行了详尽的采样,从而阐明了新的配体识别途径并发现了非天然结合姿势。










































 京公网安备 11010802027423号
京公网安备 11010802027423号