当前位置:
X-MOL 学术
›
Diam. Relat. Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ab initio description of nanodiamonds: A DFT and TDDFT benchmark
Diamond and Related Materials ( IF 4.3 ) Pub Date : 2020-10-01 , DOI: 10.1016/j.diamond.2020.107959 Diego López-Carballeira , Tomáš Polcar
Diamond and Related Materials ( IF 4.3 ) Pub Date : 2020-10-01 , DOI: 10.1016/j.diamond.2020.107959 Diego López-Carballeira , Tomáš Polcar
|
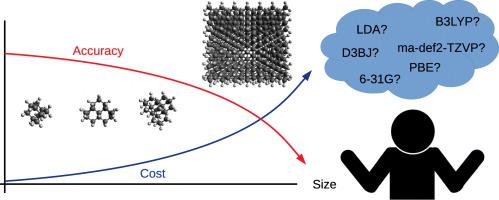
Abstract In this article 15 DFT functionals were evaluated in order to find a good description of the electronic features and geometries of the nanodiamonds. With this in mind, two sets of molecules were designed, one of them composed by well-known organic molecules and other by diamondoids. The main parameters considered for comparison in this work are the excitation energies, ionization potentials, and the energies of the frontier orbitals. Moreover, absorption distances and binding energies were also considered to assess the quality of the description of the physisorption process. The best overall performance was found on hybrid functionals and more specifically on those with low HF percentage (TPSSH and O3LYP), despite reasonable results with lower computational cost can be obtained with GGA or LDA methods. The use of a correction for the dispersion interaction is mandatory except when a Truhlar functional, LDA or ωB97X functional is used. An augmented triple zeta basis set is recommended, especially for the description of the nanodiamond model, while an augmented double zeta is enough to yield a converged geometry.
中文翻译:

纳米金刚石的从头算描述:DFT 和 TDDFT 基准
摘要 本文评估了 15 个 DFT 泛函,以更好地描述纳米金刚石的电子特征和几何形状。考虑到这一点,设计了两组分子,一组由众所周知的有机分子组成,另一组由类金刚石组成。在这项工作中考虑比较的主要参数是激发能、电离势和前沿轨道的能量。此外,还考虑了吸收距离和结合能来评估物理吸附过程的描述质量。尽管使用 GGA 或 LDA 方法可以获得具有较低计算成本的合理结果,但在混合泛函上发现了最佳的整体性能,更具体地说,在那些具有低 HF 百分比(TPSSH 和 O3LYP)的泛函上。除非使用 Truhlar 泛函、LDA 或 ωB97X 泛函,否则必须使用色散相互作用校正。推荐使用增广三 zeta 基组,特别是用于描述纳米金刚石模型,而增广双 zeta 足以产生收敛几何。
更新日期:2020-10-01
中文翻译:
纳米金刚石的从头算描述:DFT 和 TDDFT 基准
摘要 本文评估了 15 个 DFT 泛函,以更好地描述纳米金刚石的电子特征和几何形状。考虑到这一点,设计了两组分子,一组由众所周知的有机分子组成,另一组由类金刚石组成。在这项工作中考虑比较的主要参数是激发能、电离势和前沿轨道的能量。此外,还考虑了吸收距离和结合能来评估物理吸附过程的描述质量。尽管使用 GGA 或 LDA 方法可以获得具有较低计算成本的合理结果,但在混合泛函上发现了最佳的整体性能,更具体地说,在那些具有低 HF 百分比(TPSSH 和 O3LYP)的泛函上。除非使用 Truhlar 泛函、LDA 或 ωB97X 泛函,否则必须使用色散相互作用校正。推荐使用增广三 zeta 基组,特别是用于描述纳米金刚石模型,而增广双 zeta 足以产生收敛几何。









































 京公网安备 11010802027423号
京公网安备 11010802027423号