当前位置:
X-MOL 学术
›
Mol. Omics
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
LRSK: a low-rank self-representation K-means method for clustering single-cell RNA-sequencing data.
Molecular Omics ( IF 3.0 ) Pub Date : 2020-06-09 , DOI: 10.1039/d0mo00034e Ye-Sen Sun 1 , Le Ou-Yang , Dao-Qing Dai
Molecular Omics ( IF 3.0 ) Pub Date : 2020-06-09 , DOI: 10.1039/d0mo00034e Ye-Sen Sun 1 , Le Ou-Yang , Dao-Qing Dai
Affiliation
|
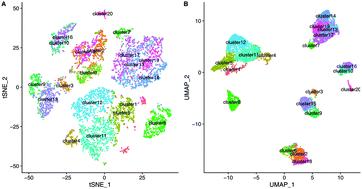
The development of single-cell RNA-sequencing (scRNA-seq) technologies brings tremendous opportunities for quantitative research and analyses at the cellular level. In particular, as a crucial task of scRNA-seq analysis, single cell clustering shines a light on natural groupings of cells to give new insights into the biological mechanisms and disease studies. However, it remains a challenge to identify cell clusters from lots of cell mixtures effectively and accurately. In this paper, we propose a novel adaptive joint clustering framework, named the low-rank self-representation K-means method (LRSK), to learn the data representation matrix and cluster indicator matrix jointly from scRNA-seq data. Specifically, instead of calculating the similarities among cells from the original data, we seek a low-rank representation of the original data to better reflect the underlying relationships among cells. Moreover, an Augmented Lagrangian Multiplier (ALM) based optimization algorithm is adopted to solve this problem. Experimental results on various scRNA-seq datasets and case studies demonstrate that our method performs better than other state-of-the-art single cell clustering algorithms. The analysis of unlabeled large single-cell liver cancer sequencing data further shows that our prediction results are more reasonable and interpretable.
中文翻译:

LRSK:一种用于对单细胞RNA测序数据进行聚类的低秩自我表示K均值方法。
单细胞RNA测序(scRNA-seq)技术的发展为细胞水平的定量研究和分析带来了巨大的机会。尤其是,作为scRNA-seq分析的一项关键任务,单细胞聚类揭示了细胞的自然分组,为生物学机制和疾病研究提供了新见识。然而,有效和准确地从大量细胞混合物中鉴定细胞簇仍然是一个挑战。在本文中,我们提出了一种新颖的自适应联合聚类框架,称为低秩自我表示K-means方法(LRSK),从scRNA-seq数据中共同学习数据表示矩阵和聚类指标矩阵。具体来说,我们不是从原始数据中计算出单元格之间的相似性,而是寻求原始数据的低等级表示,以更好地反映单元格之间的潜在关系。此外,采用基于拉格朗日乘数(ALM)的优化算法来解决此问题。在各种scRNA-seq数据集上的实验结果和案例研究表明,我们的方法比其他最新的单细胞聚类算法性能更好。对未标记的大型单细胞肝癌测序数据的分析进一步表明,我们的预测结果更加合理和可解释。
更新日期:2020-06-09
中文翻译:
LRSK:一种用于对单细胞RNA测序数据进行聚类的低秩自我表示K均值方法。
单细胞RNA测序(scRNA-seq)技术的发展为细胞水平的定量研究和分析带来了巨大的机会。尤其是,作为scRNA-seq分析的一项关键任务,单细胞聚类揭示了细胞的自然分组,为生物学机制和疾病研究提供了新见识。然而,有效和准确地从大量细胞混合物中鉴定细胞簇仍然是一个挑战。在本文中,我们提出了一种新颖的自适应联合聚类框架,称为低秩自我表示K-means方法(LRSK),从scRNA-seq数据中共同学习数据表示矩阵和聚类指标矩阵。具体来说,我们不是从原始数据中计算出单元格之间的相似性,而是寻求原始数据的低等级表示,以更好地反映单元格之间的潜在关系。此外,采用基于拉格朗日乘数(ALM)的优化算法来解决此问题。在各种scRNA-seq数据集上的实验结果和案例研究表明,我们的方法比其他最新的单细胞聚类算法性能更好。对未标记的大型单细胞肝癌测序数据的分析进一步表明,我们的预测结果更加合理和可解释。










































 京公网安备 11010802027423号
京公网安备 11010802027423号