当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Incorporating Electronic Information into Machine Learning Potential Energy Surfaces via Approaching the Ground-State Electronic Energy as a Function of Atom-Based Electronic Populations.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-06-05 , DOI: 10.1021/acs.jctc.0c00217 Xiaowei Xie 1, 2 , Kristin A Persson 2, 3 , David W Small 1, 4
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-06-05 , DOI: 10.1021/acs.jctc.0c00217 Xiaowei Xie 1, 2 , Kristin A Persson 2, 3 , David W Small 1, 4
Affiliation
|
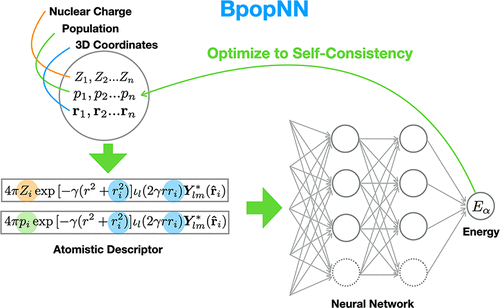
Machine learning (ML) approximations to density functional theory (DFT) potential energy surfaces (PESs) are showing great promise for reducing the computational cost of accurate molecular simulations, but at present, they are not applicable to varying electronic states, and in particular, they are not well suited for molecular systems in which the local electronic structure is sensitive to the medium to long-range electronic environment. With this issue as the focal point, we present a new machine learning approach called “BpopNN” for obtaining efficient approximations to DFT PESs. Conceptually, the methodology is based on approaching the true DFT energy as a function of electron populations on atoms; in practice, this is realized with available density functionals and constrained DFT (CDFT). The new approach creates approximations to this function with neural networks. These approximations thereby incorporate electronic information naturally into a ML approach, and optimizing the model energy with respect to populations allows the electronic terms to self-consistently adapt to the environment, as in DFT. We confirm the effectiveness of this approach with a variety of calculations on LinHn clusters.
中文翻译:

通过将基态电子能量作为基于原子的电子种群的函数,将电子信息纳入机器学习势能面。
机器学习(ML)对密度泛函理论(DFT)势能面(PESs)的近似显示了降低精确分子模拟的计算成本的巨大希望,但目前,它们不适用于变化的电子状态,尤其是,它们不适用于其中局部电子结构对中长期电子环境敏感的分子系统。以这个问题为重点,我们提出了一种新的机器学习方法,称为“ BpopNN”,用于获得DFT PES的有效近似值。从概念上讲,该方法基于逼近真正的DFT能量作为原子上电子种群的函数。实际上,这是通过可用的密度函数和受约束的DFT(CDFT)实现的。新方法使用神经网络对此函数创建了近似值。因此,这些近似值自然将电子信息合并到ML方法中,并且相对于总体优化模型能量使电子项能够如DFT中那样自洽地适应环境。我们通过对Li的各种计算来确认这种方法的有效性n H n个群集。
更新日期:2020-07-14
中文翻译:
通过将基态电子能量作为基于原子的电子种群的函数,将电子信息纳入机器学习势能面。
机器学习(ML)对密度泛函理论(DFT)势能面(PESs)的近似显示了降低精确分子模拟的计算成本的巨大希望,但目前,它们不适用于变化的电子状态,尤其是,它们不适用于其中局部电子结构对中长期电子环境敏感的分子系统。以这个问题为重点,我们提出了一种新的机器学习方法,称为“ BpopNN”,用于获得DFT PES的有效近似值。从概念上讲,该方法基于逼近真正的DFT能量作为原子上电子种群的函数。实际上,这是通过可用的密度函数和受约束的DFT(CDFT)实现的。新方法使用神经网络对此函数创建了近似值。因此,这些近似值自然将电子信息合并到ML方法中,并且相对于总体优化模型能量使电子项能够如DFT中那样自洽地适应环境。我们通过对Li的各种计算来确认这种方法的有效性n H n个群集。










































 京公网安备 11010802027423号
京公网安备 11010802027423号